
AMP-activated protein kinase
| AMP-activated protein kinase (AMPK) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
ExPASy NiceZyme view | | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
5' AMP-activated protein kinase or AMPK or 5' adenosine monophosphate-activated protein kinase is an
It should not be confused with
Structure
AMPK is a
The following human genes encode AMPK subunits:
The crystal structure of mammalian AMPK regulatory core domain (α C terminal, β C terminal, γ) has been solved in complex with AMP,[13] ADP [14] or ATP.[15]
Regulation
Due to the presence of isoforms of its components, there are 12 versions of AMPK in mammals, each of which can have different tissue localizations, and different functions under different conditions.[16] AMPK is regulated allosterically and by post-translational modification, which work together.[16]
If residue Thr-172 of AMPK's α1-subunit (or Thr-174 of AMPK's α2-subunit) is phosphorylated, AMPK is activated around 100-fold;
Regulation of AMPK by CaMKK2 requires a direct interaction of these two proteins via their kinase domains. The interaction of CaMKK2 with AMPK only involves the α and β subunits of AMPK (AMPK γ is absent from the CaMKK2 complex), thus rendering regulation of AMPK in this context to changes in calcium levels but not AMP or ADP.
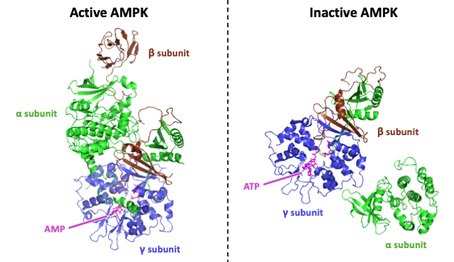
AMPK is regulated allosterically mostly by competitive binding to the CBS sites on its γ subunit between ATP (which allows phosphatase access to Thr-172) and AMP or ADP (each of which blocks access to phosphatases).[1] It thus appears that AMPK is a sensor of AMP/ATP or ADP/ATP ratios and thus cell energy level.[16] AMPK undergoes a large conformational change upon ATP binding. A region on the α subunit known as the kinase domain (KD) dissociates from its active-state conformation and loosely associates with the γ subunit ~100Å away. The KD also rotates ~180° in the conformational change. Upon KD dissociation, the active loop (AL) of the α subunit which contains the critical phosphorylated Thr residue is fully exposed to upstream phosphatases. This conformational change represents a plausible mechanism for AMPK modulation. When cellular energy states are low (high AMP/ATP or ADP/ATP levels), AMPK adopts the KD-associated conformation and AMPK is protected from dephosphorylation and remains activated. When cellular energy states are high, AMPK adopts the KD-displaced conformation, the AL is exposed to upstream phosphatases, and AMPK is deactivated.[6]
The pharmacological compounds Merck Compound 991 and Abbott A769662 bind to the allosteric drug and metabolism site (ADaM) on the β subunit and have been shown to activate AMPK up to 10-fold.[6][18] ADaM site binding may have roles in AMPK activation as well as protection against dephosphorylation.[19]
There are other mechanisms by which AMPK is inhibited or activated by insulin, leptin, and
AMPK may be inhibited or activated by various tissue-specific ubiquitinations.[16]
It is also regulated by several protein-protein interactions, and may either be activated or inhibited by oxidative factors; the role of oxidation in regulating AMPK was controversial as of 2016.[16]
Function
When AMPK phosphorylates acetyl-CoA carboxylase 1 (ACC1) or sterol regulatory element-binding protein 1c (SREBP1c), it inhibits synthesis of fatty acids, cholesterol, and triglycerides, and activates fatty acid uptake and β-oxidation.[16]
AMPK stimulates glucose uptake in skeletal muscle by phosphorylating Rab-GTPase-activating protein TBC1D1, which ultimately induces fusion of GLUT1 vesicles with the plasma membrane.[16] AMPK stimulates glycolysis by activating phosphorylation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2/3 and activating phosphorylation of glycogen phosphorylase, and it inhibits glycogen synthesis through inhibitory phosphorylation of glycogen synthase.[16] In the liver, AMPK inhibits gluconeogenesis by inhibiting transcription factors including hepatocyte nuclear factor 4 (HNF4) and CREB regulated transcription coactivator 2 (CRTC2).[16]
AMPK inhibits the energy-intensive
AMPK activates
Clinical significance
Exercise/training
Many
During a single
Mutations in the skeletal muscle calcium release channel (RYR1) underlies a life- threatening response to heat in patients with malignant hyperthermia susceptibility (MHS). Upon acute exposure to heat, these mutations cause uncontrolled Ca2+ release from the sarcoplasmic reticulum, leading to sustained muscle contractures, severe hyperthermia, and sudden death.[38] At basal conditions, the temperature-dependent Ca2+ leak also leads to increased energy demand and activation of energy sensing AMP kinase (AMPK) in skeletal muscle.[38] The activated AMPK increases muscle metabolic activity, including glycolysis, which leads to marked elevation of circulating lactate.[38]
AMPK activity increases with exercise and the LKB1/MO25/STRAD
Although AMPKα2 activation has been thought to be important for mitochondrial adaptations to exercise training, a recent study investigating the response to exercise training in AMPKα2 knockout mice opposes this idea.[45] Their study compared the response to exercise training of several proteins and enzymes in wild type and AMPKα2 knockout mice. And even though the knockout mice had lower basal markers of mitochondrial density (COX-1, CS, and HAD), these markers increased similarly to the wild type mice after exercise training. These findings are supported by another study also showing no difference in mitochondrial adaptations to exercise training between wild type and knockout mice.[46]
Maximum life span
The
Lipid metabolism
One of the effects of
Glucose transport
Two proteins are essential for the regulation of GLUT-4 expression at a transcriptional level – myocyte enhancer factor 2 (
There is another protein involved in
Mitochondria
Mitochondrial enzymes, such as
AMPK is required for increased peroxisome proliferator-activated receptor γ coactivator-1α (
To do this, it enhances the activity of
Thyroid hormone
AMPK and
Glucose sensing systems
Loss of AMPK has been reported to alter the sensitivity of glucose sensing cells, through poorly defined mechanisms. Loss of the AMPKα2 subunit in pancreatic β-cells and hypothalamic neurons decreases the sensitivity of these cells to changes in extracellular glucose concentration.[58][59][60][61] Moreover, exposure of rats to recurrent bouts of insulin induced hypoglycemia/glucopenia, reduces the activation of AMPK within the hypothalamus, whilst also suppressing the counterregulatory response to hypoglycemia. [62][63] Pharmacological activation of AMPK by delivery of AMPK activating drug AICAR, directly into the hypothalamus can increase the counterregulatory response to hypoglycaemia.[64]
Lysosomal damage, inflammatory diseases and metformin
AMPK is recruited to lysosomes and regulated at the lysosomes via several systems of clinical significance. This includes the
Tumor suppression and promotion
Some evidence indicates that AMPK may have a role in tumor suppression. Studies have found that AMPK may exert most, or even all of, the tumor suppressing properties of liver kinase B1 (LKB1).[17] Additionally, studies where the AMPK activator metformin was used to treat diabetes found a correlation with a reduced risk of cancer, compared to other medications. Gene knockout and knockdown studies with mice found that mice without the gene to express AMPK had greater risks of developing lymphomas, though as the gene was knocked out globally instead of just in B cells, it was impossible to conclude that AMP knockout had cell-autonomous effects within tumor progenitor cells.[73]
In contrast, some studies have linked AMPK with a role as a tumor promoter by protecting cancer cells from stress. Thus, once cancerous cells have formed in an organism, AMPK may swap from protecting against cancer to protecting the cancer itself. Studies have found that tumor cells with AMPK knockout are more susceptible to death by glucose starvation or extracellular matrix detachment, which may indicate AMPK has a role in preventing these two outcomes. There is no direct evidence that inhibiting AMPK would be an effective cancer treatment in humans.[73]
Controversy over role in adaption to exercise/training
This section is written like a personal reflection, personal essay, or argumentative essay that states a Wikipedia editor's personal feelings or presents an original argument about a topic. (January 2021) |
A seemingly
Because the AMPK response to exercise decreases with increased training duration, many questions arise that would challenge the AMPK role with respect to
If the AMPK response to exercise is responsible in part for biochemical adaptations to training, how then can these adaptations to training be maintained if the AMPK response to exercise is being attenuated with training? It is hypothesized that these adaptive roles to training are maintained by AMPK activity and that the increases in AMPK activity in response to exercise in trained skeletal muscle have not yet been observed due to biochemical adaptations that the training itself stimulated in the
See also
Notes
- ^ Leptin is secreted by adipose tissue upon insulin stimulus, and it inhibits AMPk in hypothalamus (reducing appetite) but stimulates AMPk in peripheral tissues.
References
- ^ PMID 16943194.
- PMID 10409121.
- PMID 19211918.
- ^ PMID 8557660.
- PMID 18372250.
- ^ PMID 34437114.
- PMID 20974912.
- S2CID 13591617.
- ^ PMID 14691231.
- ^ PMID 8910387.
- PMID 9575201.
- PMID 10698692.
- S2CID 4345919.
- PMID 21399626.
- S2CID 13591617.
- ^ PMID 27416781.
- ^ PMID 14614828.
- PMID 25066137.
- PMID 24352254.
- PMID 22436748.
- ^ S2CID 21577702.
- ^ PMID 12444247.
- ^ S2CID 9622267.
- ^ S2CID 7891060.
- ^ S2CID 27188844.
- ^ PMID 15294043.
- ^ S2CID 5627995.
- ^ PMID 15790954.
- ^ PMID 9703344.
- ^ PMID 10871188.
- ^ S2CID 29891424.
- ^ PMID 9435525.
- ^ PMID 11069105.
- ^ PMID 16364253.
- ^ S2CID 17877895.
- ^ S2CID 22504819.
- PMID 18674809.
- ^ PMID 33037202.
- PMID 10642499.
- PMID 14511394.
- S2CID 25296834.
- PMID 9124333.
- ^ PMID 15292028.
- ^ S2CID 14454264.
- PMID 16954334.
- PMID 17513699.
- PMID 17908557.
- S2CID 18290292.
- S2CID 4409274.
- ^ PMID 15068958.
- S2CID 4302317.
- PMID 15644453.
- S2CID 39876683.
- PMID 16926377.
- ^ PMID 12433937.
- S2CID 2019764.
- PMID 12546681.
- PMID 20221584.
- PMID 20465544.
- PMID 17671657.
- PMID 22760787.
- PMID 17185376.
- PMID 17977955.
- PMID 19357294.
- PMID 31204282.
- ^ PMID 31995728.
- PMID 27753622.
- ^ PMID 31813797.
- PMID 23412688.
- PMID 27693506.
- PMID 30185776.
- PMID 27732831.
- ^ .
External links
- AMP-activated+protein+kinase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- EC 2.7.11.31