
Acetyl-CoA carboxylase
| acetyl-CoA carboxylase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
ExPASy NiceZyme view | | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
Chr. 17 q21 | |||||||
|---|---|---|---|---|---|---|---|
| |||||||
Chr. 12 q24.1 | |||||||
|---|---|---|---|---|---|---|---|
| |||||||
Acetyl-CoA carboxylase (ACC) is a
Structure
In mammals where two isoforms of ACC are expressed, the main structural difference between these isoforms is the extended ACC2 N-terminus containing a
-
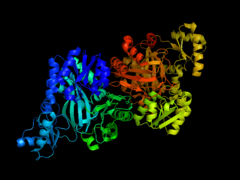 Biotin carboxylase subunit of E. coli acetyl-CoA carboxylase
Biotin carboxylase subunit of E. coli acetyl-CoA carboxylase -
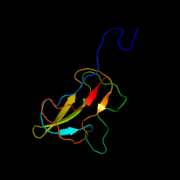 Biotin carboxyl carrier protein subunit of E. coli acetyl-CoA carboxylase
Biotin carboxyl carrier protein subunit of E. coli acetyl-CoA carboxylase -
 Carboxyl transferase subunit of E. coli acetyl-CoA carboxylase
Carboxyl transferase subunit of E. coli acetyl-CoA carboxylase



Genes
The polypeptides composing the multi-subunit ACCs of
Mechanism
The overall reaction of ACAC(A,B) proceeds by a two-step mechanism.

In the
Function
The function of ACC is to regulate the metabolism of fatty acids. When the enzyme is active, the product, malonyl-CoA, is produced which is a building block for new fatty acids and can inhibit the transfer of the fatty acyl group from acyl CoA to
In
A mitochondrial isoform of ACC1 (mACC1) plays a partially redundant role in lipoic acid synthesis and thus in protein lipoylation by providing malonyl-CoA for mitochondrial fatty acid synthesis (mtFASII) in tandem with ACSF3.[12][13]
Regulation

The regulation of mammalian ACC is complex, in order to control two distinct pools of malonyl CoA that direct either the inhibition of beta oxidation or the activation of lipid biosynthesis.[14]
Mammalian ACC1 and ACC2 are regulated transcriptionally by multiple
Through a feed-forward loop,
Phosphorylation can result when the hormones
When
This protein may use the morpheein model of allosteric regulation.[26]
Clinical implications
At the juncture of lipid synthesis and oxidation pathways, ACC presents many clinical possibilities for the production of novel
In addition, plant-selective ACC inhibitors are in widespread use as herbicides,[32] which suggests clinical application against Apicomplexa parasites that rely on a plant-derived ACC isoform,[33] including malaria.
The heterogeneous clinical phenotypes of the metabolic disease combined malonic and methylmalonic aciduria (CMAMMA) due to ACSF3 deficiency are thought to result from partial compensation of a mitochondrial isoform of ACC1 (mACC1) for deficient ACSF3 in mitochondrial fatty acid synthesis (mtFASII).[34]
See also
References
- ^ S2CID 1131957.
- PMID 9449982.
- PMID 7732023.
- PMID 8670171.
- S2CID 41506311.
- ^ "accA, acetyl-CoA carboxylase alpha subunit (Escherichia coli str. K-12 substr. MG1655)". NCBI gene. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "accD, acetyl-CoA carboxylase beta subunit (Escherichia coli str. K-12 substr. MG1655)". NCBI gene. National Center for Biotechnology Information, U.S. National Library of Medicine.
- S2CID 24548083.
- ^ PMID 19213731.
- PMID 8753813.
- ^ PMID 15749055.
- PMID 28986507.
- S2CID 220472402.
- ^ S2CID 748630.
- PMID 12213084.
- PMID 15496471.
- PMID 14470343.
- PMID 10753875.
- PMID 9173866.
- PMID 35608294.
- S2CID 81688050.
- PMID 17253964.
- PMID 12015362.
- S2CID 52193976.
- PMID 16545081.
- PMID 22182754.
- PMID 18221116.
- PMID 16103361.
- PMID 26976583.
- ^ Tong A (11 April 2019). "Gilead shores up hope for NASH cocktail with a glimpse at positive proof-of-concept data". Endpoints News.
- PMID 30324141.
- ^ Al-Khatib K. "Acetyl CoA Carboxylase (ACCase) Inhibitors". Herbicide Symptoms. Division of Agriculture and Natural Resources, University of California, Davis.
- PMID 10557330.
- PMID 31969167.
Further reading
- Voet D, Voet JG (2004). Biochemistry (3rd ed.). Wiley. ISBN 978-0-471-19350-0.
- Buchanan BB, Gruissem W, Jones RL, eds. (2000). Biochemistry and molecular biology of plants. American Society of Plant Physiologists. ISBN 978-0-943088-37-2.
- Levert KL, Waldrop GL, Stephens JM (May 2002). "A biotin analog inhibits acetyl-CoA carboxylase activity and adipogenesis". The Journal of Biological Chemistry. 277 (19): 16347–16350. PMID 11907024.