
Atypical teratoid rhabdoid tumor
| Atypical teratoid rhabdoid tumor | |
|---|---|
 | |
| MRI of an AT/RT | |
| Specialty | Neuro-oncology |
| Usual onset | Age 3 and younger[1] |
| Prognosis | Five-year survival rate: 32.2%[2] |
| Frequency | ~58 new diagnoses per year (United States)[2] |
An atypical teratoid rhabdoid tumor (AT/RT) is a rare
In the United States, three children per 1,000,000 or around 30 new AT/RT cases are diagnosed each year. AT/RT represents around 3% of pediatric cancers of the CNS.[4] Around 17% of all pediatric cancers involve the CNS, making these cancers the most common childhood solid tumor.[citation needed] The survival rate for CNS tumors is around 60%. Pediatric brain cancer is the second-leading cause of childhood cancer death, just after leukemia. Recent trends suggest that the rate of overall CNS tumor diagnosis is increasing by about 2.7% per year. As diagnostic techniques using genetic markers improve and are used more often, the proportion of AT/RT diagnoses is expected to increase.
AT/RT was only recognized as an entity in 1996 and added to the World Health Organization Brain Tumor Classification in 2000 (Grade IV).[5] The relatively recent classification and rarity has contributed to initial misdiagnosis and nonoptimal therapy. This has led to a historically poor prognosis.[6]
Current research is focusing on using chemotherapy protocols that are effective against rhabdomyosarcoma in combination with surgery and radiation therapy.
Recent studies using multimodal therapy have shown significantly improved survival data. In 2008, the Dana-Farber Cancer Institute in Boston reported two-year overall survival of 53% and event-free survival of 70% (median age at diagnosis of 26 months).[7] In 2013, the Medical University of Vienna reported five-year overall survival of 100%, and event-free survival of 89% (median age at diagnosis of 24 months).[8]
Survival rates can be significantly improved when the correct genetic diagnosis is made at the outset, followed with specific multimodal treatment.
Signs and symptoms
Clinical signs and symptoms depend on the location of the tumor.
Since many of the tumors occur in the
Genetics
The rate of
Risk for siblings and other members of the family
Atypical teratoid/rhabdoid tumors are very rare, and absolute risk to siblings is not reported in the literature. However, some reports exist of AT/RTs presenting in two members of the same family, or one family member with an AT/RT and another with a renal rhabdoid tumor or other CNS tumor. These are suspected to arise from germline genetic mutations in a parent shared by affected siblings.
- A three-generation family is known in which two half-brothers were diagnosed with CNS atypical teratoid/rhabdoid tumors (AT/RT). The two boys, diagnosed at 2 months and 17 months of age, had a germline insertion mutation in exon 4 of the INI1 gene that was inherited from their healthy mother. A maternal uncle died in childhood from a brain tumor and a malignant rhabdoid tumor of the kidney. The identification of two unaffected carriers in a family segregating a germline mutation and rhabdoid tumor supports the hypothesis that variable risks of development of rhabdoid tumor in the context of a germline mutation may exist. Most rhabdoid tumors may occur in a developmental window. This family highlights the importance of mutation analysis in all patients with a suspected rhabdoid tumor.[11]
- In the first case report of monozygotic twins, both with brain tumors having similar genetic alterations, authors suggest a common genetic pathway.[12]
- A case was reported of an infant who developed both AT/RT and renal rhabdoid tumors that were identical in gross and immunologic histology.[13]
- A family has had multiple generations of posterior fossa tumors including rhabdoid tumors and choroid plexus carcinoma. A germline mutation (SMARCB1) was found in both affected and some unaffected family members.[14]
- Two sisters were diagnosed with AT/RTs 15 days apart. A case report stated no karyotypic anomalies were noted.[15]
- Three siblings had a mutation of the SMARCB1 gene and one had a choroid plexus carcinoma and two had an AT/RT. Although the mother had a normal somatic DNA, the mutation apparently was inherited from the mother's germline due to a mutation during oogenesis.[16]
- Izycka-Swieszewska et al. describe a five-month-old child with an AT/RT, whose father was diagnosed with a primitive neuroectodermal tumor (PNET) of the spinal canal. Fluorescent in situ hybridization analysis showed significant genetic differences in the specimens which suggest that the occurrence of these virulent CNS malignancies within a single family was coincidental.[17]
Pathology
AT/RT and rhabdoid tumor share the term "rhabdoid" because under a microscope, both tumors resemble rhabdomyosarcoma[citation needed].
-
 AT/RT Histology with numerous rhabdoid tumor cells
AT/RT Histology with numerous rhabdoid tumor cells -
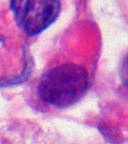 Rhabdoid Tumor Cell - 400X Magnification
Rhabdoid Tumor Cell - 400X Magnification


Histology
The tumor
Immunohistochemistry

Immunohistochemical staining is widely used in the diagnosis and treatment of cancer. Specific molecular markers are characteristic of particular cancer types. Immunohistochemistry is also widely used in basic research to understand the distribution and localization of biomarkers in different parts of a tissue. Proteins found in an ATeratoid/RT are:[citation needed]
- Vimentin-positive
- Cytokeratin-positive
- Neuron specific enolase-positive
- Epitelial membrane antigen-positive
- Glial fibrillary acidic protein- positive
- Synaptophysin
- Chromogranin
- Smooth muscle actin
- Desmin
- Carcinoembrionary antigen
- CD99 antigen;[19][20]
- S-100
- neurofilaments
- AFP – not found
- HCG – negative
Cytogenetic studies
In importance of the hSNF5/INI1 gene located on chromosomal band 22q11.2 is highlighted, as the mutation's presence is sufficient to change the diagnosis from a medulloblastoma or PNET to the more aggressive AT/RT classification. However, this mutation is not present in 100% of cases. Therefore, if the mutation is not present in an otherwise classic AT/RT immunohistochemical and morphologic pattern then the diagnosis remains an AT/RT.
Diagnosis
The standard work-up for AT/RT includes:[citation needed]
- Magnetic resonance imaging (MRI) of the brain and spine
- Lumbar puncture to look for M1 disease
- Computed tomography(CT) of chest and abdomen to check for a tumor
- Bone marrow aspiration to check for bone tumors. Sometimes the physician will perform a stem cell transplant
- Bone marrow biopsy
- Bone scan
The initial diagnosis of a tumor is made with a
Examination of the cerebrospinal fluid is important (CSF), as one-third of patients will have intracranial dissemination with involvement of the CSF. Large tumor cells, eccentricity of the nuclei, and prominent nucleoli are consistent findings.[23] Usually only a minority of AT/RT biopsies have rhabdoid cells, making diagnosis more difficult. Increasingly it is recommended that a genetic analysis be performed on the brain tumor, especially to find if a deletion in the INI1/hSNF5 gene is involved (appears to account for over 80% of the cases). The correct diagnosis of the tumor is critical to any protocol. Studies have shown that 8% to over 50% of AT/RT tumors are diagnosed incorrectly.[citation needed]
Classification
AT/RT may be related to
Differential diagnosis
The critical step in treatment planning is to determine the correct histology of the tumor. Misidentification of the tumor histology can lead to errors in treatment and prognosis.[24]
Atypical teratoid/rhaboid tumor closely resembles medulloblastoma,[25] primitive neuroectodermal tumor, choroid plexus carcinoma, and some kinds of germ cell tumor.[citation needed] Because rhabdoid characteristics are not the only component of AT/RT, some sections of an AT/RT may resemble other tumors. These characteristics may be present only in focal areas or may be less pronounced.[citation needed]
Consideration of AT/RT when a medulloblastoma or PNET is suspected is important, particularly in a child under the age of one.
Appearance on radiologic exam
AT/RTs can occur at any sites within the CNS; however, about 60% are located in the posterior fossa or cerebellar area. The ASCO study showed 52% posterior fossa; 39% sPNET; 5% pineal; 2% spinal, and 2% multifocal.[3]
The tumors' appearance on CT and MRI are not specific, tending towards large size, calcifications, necrosis (tissue death), and hemorrhage (bleeding). Radiological studies alone cannot identify AT/RT; a pathologist almost always has to evaluate a brain tissue sample.
The increased cellularity of the tumor may make the appearance on an uncontrasted CT to have increased attenuation. Solid parts of the tumor often enhance with contrast MRI finding on T1 and T2 weighted images are variable. Precontrast T2 weighted images may show an isosignal or slightly hypersignal. Solid components of the tumor may enhance with contrast, but not always. MRI studies appear to be more able to pick up metastatic foci in other intracranial locations, as well as intraspinal locations.
Preoperative and follow-up studies are needed to detect metastatic disease.
Treatment
Surgery
Surgery plays a critical role in obtaining tissue to make an accurate diagnosis. Surgery alone is not curative. In addition, 30% of the AT/RTs are located supratentorially and a predilection exists for the cerebellopontine angle,[26] which makes surgical resection difficult. One-third or more children will have disseminated disease at the time of diagnosis. Total or near-total resections are often not possible.
Chemotherapy
Around 50% of the AT/RTs will transiently respond, but
are listed below:- CCG clinical trial CCG-9921 was activated in 1993 and published its results in 2005. The proposed treatments did not have different outcomes and were not an improvement on prior treatments.[27] Geyer published a review of chemotherapy on 299 infants with CNS tumors that evaluated response rate, event-free survival (EFS), and toxicity of two chemotherapeutic regimens for treatment of children younger than 36 months with malignant brain tumors. Patients were randomly assigned to one of two regimens of induction chemotherapy (vincristine, cisplatin, cyclophosphamide, and etoposide v vincristine, carboplatin, ifosfamide, and etoposide). Intensified induction chemotherapy resulted in a high response rate of malignant brain tumors in infants. Survival was comparable to that of previous studies, and most patients who survived did not receive radiation therapy.[27]
- intrathecal chemotherapy similar to the Intergroup Rhabdomyosarcoma Study III guidelines.[28]
- Protocol uses intrathecal mafosfamide, a pre-activated cyclophosphamide derivative, in addition to other modalities to try to effect this tumor.[29]
- High dose chemotherapy with stem cell rescue. This therapy uses chemotherapy at doses high enough to completely suppress the bone marrow transplantation, was initially thought to be of benefit to a wide group of patients, but has declined over the history of chemotherapyprotocols.
Radiation therapy
The traditional practice for childhood brain tumors has been to use chemotherapy and to defer radiation therapy until a child is older than three years. This strategy is based upon observations that children under three have significant long-term complications as a result of brain irradiation. However, the long-term outcomes of AT/RT are so poor that some protocols call for upfront radiation therapy, often in spite of young age.[30]
The dose and volume of radiation had not been standardized, but radiation does appear to improve survival. The use of radiation has been limited in children younger than three because of the risk of severe neurocognitive deficits. Protocols using conformal, local radiation in the young child are used to try to cure this tumor.
External beam (conformal) radiation uses several beams that intersect at the tumor location; the normal brain tissue receives less radiation and cognitive function is thereby less affected.
Proton beam radiation was only offered at Massachusetts General Hospital in Boston and at Loma Linda, California, as of 2002. Since 2003, three or four more proton therapy centers have opened in the United States. St. Jude Children's Research Hospital is in the process of building one at their Memphis, Tennessee, location. Some centers have since opened in Europe. (Germany, Switzerland, and France).[31][32][33][34][35][36]
Chromatin remodeling agents
This protocol is still in preclinical evaluation. Histone deacetylase inhibitors are a new class of anticancer agents targeted directly at chromatin remodeling. These agents have been used in acute promyelocytic leukemia and have been found to affect the HDAC-mediated transcriptional repression. Understanding of the INI1 deficiency is insufficient to predict whether HDAC inhibitors will be effective against AT/RTs. Some laboratory results indicate it is effective against certain AT/RT cell lines.[37]
Prognosis
The prognosis for AT/RT has been very poor, although some indications exist that an IRSIII-based therapy can produce long-term survival (60 to 72 months). Two-year survival is less than 20%, average survival postoperatively is 11 months, and doctors often recommend palliative care, especially with younger children because of the poor outcomes.[citation needed] Recently, a protocol used by a multicenter trial reported in the Journal of Clinical Oncology resulted in a 70% survival rate at 2–3 years, with most relapses occurring within months, leading to hope that a point exists beyond which patients can be considered cured.[38]
Patients with metastasis (disseminated tumor), larger tumors, tumors that could not be fully removed, or tumor recurrence, and who were younger than 36 months had the worst outcomes (i.e., shorter survival times).[citation needed]
A retrospective survey from 36 AT/RT cases at
The longest-term survivals reported in the literature are:
- (a) Hilden and associates reported a child who was still free from disease at 46 months from diagnosis.[39]
- (b) Olson and associates reported a child who was disease free at five years from diagnosis based on the IRS III protocol.[40]
- (c) In 2003, Hirth reported a patient who had been disease-free over six years.[41]
- (d) Zimmerman in 2005 reported 50-to-72 month survival rates on four patients using an IRS III-based protocol. Two of these long-term survivors had been treated after an AT/RT recurrence.[42]
- (e) A NYU study (Gardner 2004) has four of 12 longer-term AT/RT survivors; the oldest was alive at 46 months after diagnosis.[43]
- (f) Medical University of Vienna, 2013, reported a 16-year survivor, among other long-term survivors [8]
Cancer treatments in long-term survivors who are children usually cause a series of negative effects on physical well-being, fertility, cognition, and learning.[44][45][46][47]
Metastasis
Metastatic spread is noted in roughly one-third of the AT/RT cases at the time of diagnosis, and tumors can occur anywhere throughout the CNS. The ASCO study of the 188 documented AT/RT cases prior to 2004 found 30% of the cases had metastasis at diagnosis.[3] Metastatic spread to the meninges (leptomenigeal spread sometimes referred to as sugar coating) is common both initially and with relapse. Average survival times decline with the presence of metastasis. Primary CNS tumors generally metastasize only within the CNS.
One case of metastatic disease to the abdomen via ventriculoperitoneal shunt has been reported with AT/RT . Metastatic dissemination via this mechanism has been reported with other brain tumors, including germinomas, medulloblastomas, astrocytomas, glioblastomas, ependymomas, and endodermal sinus tumors. Guler and Sugita separately reported cases of lung metastasis without a shunt.[48][49]
Epidemiology
An estimated 3% of pediatric brain tumors are AT/RTs, although this percentage may increase with better differentiation between PNET/medulloblastoma tumors and AT/RTs.[citation needed]
As with other CNS tumors, more males are affected than females (ratio 1.6:1). The ASCO study showed a 1.4:1 male to female ratio.[3]
History
Atypical teratoid/rhabdoid tumor was first described as a distinct entity in 1987.[
By 1995, AT/RT had become regarded as a newly defined aggressive, biologically unique class of primarily brain and spinal tumors, usually affecting infants and young children.
Research directions
Atypical teratoid rhabdoid tumor is rare, and no therapy has been proven to deliver long-term survival, nor a set of protocols made standard. Thus, most children with AT/RT are enrolled in clinical trials to attempt to find an effective cure. A clinical trial is not a treatment standard; it is research. Some clinical trials compare an experimental treatment to a standard treatment, but only if such a standard treatment exists.
Research into stem cell transplant surgeries is ongoing.
Society and culture
In 2011, The New Yorker published an article by Aleksandar Hemon, about the author's daughter's battle with AT/RT.[52]
In August 2011, a 6-year-old named Avalanna Routh who was battling AT/RT at Dana–Farber Cancer Institute was given a pretend wedding with her idol Justin Bieber, with doctors and nurses providing a cardboard life-sized cutout of Bieber, a guitarist, flowers, and a T-shirt emblazoned with the words "Future Mrs. Bieber". In February 2012, she spent the day in person with Justin Bieber, her pretend husband, after a Facebook campaign to meet her idol.[53] On September 26, 2012, she died after battling AT/RT for five and a half years.[54]
The video game
See also
References
- ^ "Atypical Teratoid Rhabdoid Tumor (ATRT)". St. Jude Children's Research Hospital. Retrieved Mar 8, 2023.
- ^ a b "Atypical Teratoid Rhabdoid Tumor (ATRT) Diagnosis and Treatment". National Cancer Institute. Retrieved Mar 8, 2023.
- ^ a b c d Kieran MW (2006). "An Update on Germ Cell Tumors, Atypical Teratoid/Rhaboid Tumors, and Choroid Plexus Tumors Rare Tumors 3: Brain Tumors---Germ Cell Tumors, Atypical Teratoid/Rhabdoid Tumors, and Choroid Plexus Tumors". American Society of Clinical Oncology. Education. Book. Archived from the original on 2008-01-07. Retrieved 2007-05-20.
- ^ Measure D6: Types of Childhood Cancer – 2006 Tables D6a & D6b. U.S. Environmental Protection Agency. Retrieved on 2008-04-17.
- ISBN 92-83-22409-4.
- ^ PMID 15735125. See Figure 1.
- PMID 19064966.
- ^ PMID 24402832.
- ^
Tamiya T, Nakashima H, Ono Y, Kawada S, Hamazaki S, Furuta T, et al. (March 2000). "Spinal atypical teratoid/rhabdoid tumor in an infant". Pediatric Neurosurgery. 32 (3): 145–149. S2CID 34074177.
- ^
Kao CL, Chiou SH, Chen YJ, Singh S, Lin HT, Liu RS, et al. (June 2005). "Increased expression of osteopontin gene in atypical teratoid/rhabdoid tumor of the central nervous system". Modern Pathology. 18 (6): 769–778. PMID 15776015.
- S2CID 25821741.
- S2CID 45546297.
- S2CID 21250260.
- PMID 10739763.
- S2CID 10500489.
- PMID 10521299.
- S2CID 2766929.
- S2CID 22159380.
- ^ "CD99". Ncbi.nlm.nih.gov. 2013-01-30. Retrieved 2013-02-22.
- ^ "2nd CD99 link". Ncbi.nlm.nih.gov. Retrieved 2013-02-22.
- PMID 11230702.
- ^
Meyers SP, Khademian ZP, Biegel JA, Chuang SH, Korones DN, Zimmerman RA (May 2006). "Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes". AJNR. American Journal of Neuroradiology. 27 (5): 962–971. PMID 16687525. Retrieved 2008-05-05.
- S2CID 21276083.
- PMID 9185218.
- ^
Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, et al. (September 1998). "Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study". The American Journal of Surgical Pathology. 22 (9): 1083–1092. PMID 9737241.
- ^ "PDF" (PDF). Archived from the original (PDF) on 2013-02-17. Retrieved 2013-02-22.
- ^ PMID 16234523.
- ^ "Childhood Rhabdomyosarcoma Treatment". National Cancer Institute. 1980-01-01. Archived from the original on 4 July 2007. Retrieved 2007-07-09.
- PMID 17416804.
- S2CID 22170046.
- ^ "Principles of Proton Beam Therapy". Massgeneral.org. Archived from the original on 2013-09-26. Retrieved 2013-02-22.
- ^ Owen C. "Proton Beam Radiotherapy at Mass. General". Neurosurgery.mgh.harvard.edu. Archived from the original on 2012-11-12. Retrieved 2013-02-22.
- ^ Proton Beam Therapy Article Archived October 8, 2007, at the Wayback Machine
- PMID 16189526.
- ^ Loma Linda Medical Center Proton Treatment Center - Overview Archived February 6, 2007, at the Wayback Machine
- ^ Loma Linda overview of Childhood Brain Tumors Archived April 12, 2007, at the Wayback Machine
- PMID 12138206.
- ^ "New hope for patients with rare brain tumors". Danafarberchildrens.org. 2009-03-10. Retrieved 2013-02-22.
- ^ PMID 15254056.
- S2CID 1872438.
- .
- S2CID 22624213.
- S2CID 1844660.
- ^
Fouladi M, Gilger E, Kocak M, Wallace D, Buchanan G, Reeves C, et al. (October 2005). "Intellectual and functional outcome of children 3 years old or younger who have CNS malignancies". Journal of Clinical Oncology. 23 (28): 7152–7160. PMID 16192599.
- ^ Monteleone P, Meadows AT (June 6, 2006). "Late Effects of Childhood Cancer and Treatment". EMedicine from WebMD.
- ^
Foreman NK, Faestel PM, Pearson J, Disabato J, Poole M, Wilkening G, et al. (January 1999). "Health status in 52 long-term survivors of pediatric brain tumors". Journal of Neuro-Oncology. 41 (1): 47–53. S2CID 10230653.
- ^
Meyer EA, Kieran MW (2002). "Psychological adjustment of 'surgery-only' pediatric neuro-oncology patients: a retrospective analysis". Psycho-Oncology. 11 (1): 74–79. S2CID 21473233. Archived from the originalon 2012-12-17.
- S2CID 349180.
- S2CID 24672683.
- S2CID 8100347.
- PMID 11782395.
- ^ Hemon A (June 13, 2011). "The Aquarium". The New Yorker. Retrieved March 2, 2012.
- ^ "Justin Bieber Meets With Merrimac Girl Battling Brain Cancer". CBS Boston. CBS Local Media, a division of CBS Radio Inc. February 13, 2012. Retrieved March 2, 2012.
- ^ "'Mrs. Bieber' Avalanna Routh dies at age 6". CNN.com. September 27, 2012. Archived from the original on October 23, 2012. Retrieved October 26, 2012.
- ^ Robertson A (July 12, 2013). "That Dragon, Cancer: the video game helping a father face his son's disease". The Daily Telegraph. Retrieved March 13, 2014.
- ^ Futter M (March 13, 2014). "Joel Green, Inspiration For That Dragon, Cancer, Passes Away At Age 5". Game Informer. Retrieved March 13, 2014.
Further reading
- Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, et al. (July 2004). "Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry". Journal of Clinical Oncology. 22 (14): 2877–2884. PMID 15254056.
- Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, et al. (January 2002). "The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors". Cancer Research. 62 (1): 323–328. PMID 11782395.
- Huret J, Sevenet N (2002). "Rhabdoid predispostion syndrome". Atlas of Genetics and Cytogenetics in Oncology and Haematology. Archived from the original on 2005-12-26.