
Mukaiyama hydration
The Mukaiyama hydration is an
The reaction was developed by Teruaki Mukaiyama at Mitsui Petrochemical Industries, Ltd. Its discovery was based on previous work on the selective hydrations of olefins catalyzed by cobalt complexes with Schiff base ligands[2] and porphyrin ligands.[3] Due to its chemoselectivity (tolerant of other functional groups) and mild reactions conditions (run under air at room temperature), the Mukaiyama hydration has become a valuable tool in chemical synthesis.
Mechanism
In his original publication, Mukaiyama proposed that the reaction proceeded through the intermediacy of a cobalt peroxide adduct. A metal exchange reaction between a hydrosilane and the cobalt peroxide adduct leads to a silyl peroxide, which is converted to the alcohol upon reduction, presumably via action of the cobalt catalyst.

Studies investigating the mechanism of cobalt-catalyzed peroxidation of alkenes by Nojima and coworkers,[4] support the intermediacy of a metal hydride that reacts with the alkene directly to form a transient cobalt-alkyl bond. Homolysis generates a carbon centered radical that reacts directly with oxygen and is subsequently trapped by a cobalt(II) species to form the same cobalt-peroxide adduct as suggested by Mukaiyama. Metal exchange with the hydrosilane produces a silyl peroxide product and further reduction (via homolysis of the oxygen-oxygen bond) leads to the product alcohol. The use of a silane reductant allows for this reaction to be carried out without heat.[5] The authors also note, in accordance with previous studies,[6] that the addition of t-butylhydroperoxide can increase the rate of slower-reacting substrates. This rate increase is likely due to oxidation of cobalt(II) to alkylperoxo-cobalt(III) complex, which subsequently participates in a rapid metal exchange with the hydrosilane to generate the active cobalt(III)-hydride.

It is important to note that the mechanism laid out above is in marked contrast to previous mechanistic proposals,[7] which suggest that a cobalt-peroxy complex inserts directly into alkenes. The aforementioned study by Nojima and coworkers disagrees with this proposal due to three observations: 1) the intermediacy of a cobalt-hydride observed via 1H NMR 2) the propensity of alkenes to undergo autooxidation to the α, β-unsaturated ketones or allylic alcohols when the same reaction is run in the absence of a hydrosilane 3) the predominant mode of decomposition of alkylperoxo-cobalt(III) species to an alkoxy or alkylperoxy radical via the Haber–Weiss mechanism.
A recent review by Shenvi and coworkers,[8] proposed that the Mukaiyama hydration operates via the same principles as metal hydride hydrogen atom transfer (MH HAT), elucidated by Jack Halpern and Jack R. Norton in their studies on hydrogenation of anthracenes by syngas and Co2(CO)8[9] and the chemistry of vitamin B12 mimics,[10] respectively.
Variations
Carbon-oxygen bond formation
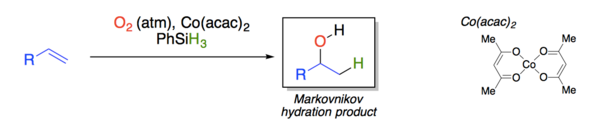
Yamada explored the effect of different solvents and cobalt beta-diketonate ligands on the yield and product distribution of the reaction.[11]

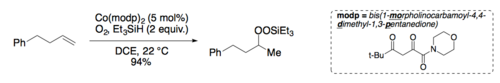
Mukaiyama and Isayama developed conditions to isolate the intermediate silylperoxide.[6][12] Treatment of the intermediate silylperoxide with 1 drop of concentrated HCl in methanol leads to the hydroperoxide product.
Both Mukaiyama[13] and Magnus[14] describe conditions for an α-enone hydroxylation reaction using Mn(dpm)x in the presence of oxygen and phenylsilane. An asymmetric variant was described by Yamada and coworkers.[15]

Carbon-nitrogen bond formation
Erick Carreira’s group has developed both cobalt and manganese-catalyzed methods for the hydrohydrazination of olefins.[17][18]

Both Carreira[19] and Boger[20] have developed hydroazidation reactions.
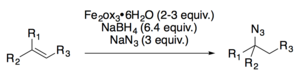
Applications
In total synthesis
The Mukaiyama hydration or variants thereof have been featured in the syntheses of (±)-garsubellin A,[21] stigmalone,[22] vinblastine,[23] (±)-cortistatin A,[24] (±)-lahadinine B,[25] ouabagenin,[26] pectenotoxin-2,[27] (±)-indoxamycin B,[28] trichodermatide A,[29] (+)-omphadiol[30] and many more natural products.
In the following diagram, an application of the Mukaiyama hydration in the total synthesis of (±)-garsubellin A is illustrated:
-Garsubellin_A.svg)
The hydration reaction is catalyzed by Co(acac)2 (acac = 2,4-pentanedionato, better known as acetylacetonato) and carried out in the presence of air oxygen & phenylsilane. With isopropanol used as solvent, yields of 73 % are obtained.
See also
- Hydration reaction
- Oxymercuration-reduction
- Hydroboration-oxidation reaction
References
- ISSN 0366-7022.
- ISSN 0002-7863.
- ISSN 0022-3263.
- PMID 12375896.
- PMID 28525261.
- ^ ISSN 0366-7022.
- .
- PMID 27461578.
- S2CID 53710591.
- PMID 25427140.
- .
- ISSN 0009-2673.
- ISSN 0366-7022.
- .
- ISSN 0366-7022.
- PMID 19292450.
- PMID 16146399.
- PMID 15300706.
- PMID 15941257.
- PMID 22369097.
- PMID 16218611.
- S2CID 196825043.
- PMID 19292450.
- PMID 18479104.
- .
- PMID 23288535.
- PMID 15844913.
- PMID 22345071.
- PMID 23417860.
- PMID 21761524.