
T-cell receptor
| TCR complex | |
|---|---|
CD247) accessory molecules | |
| Identifiers | |
| Symbol | TCR |
| OPM superfamily | 166 |
| Membranome | 26 |

| T-cell receptor alpha locus | |
|---|---|
| Identifiers | |
| Symbol | Chr. 14 q11.2 |
| T-cell receptor beta locus | |
|---|---|
| Identifiers | |
| Symbol | TRB |
| Alt. symbols | TCRB, TRB@ |
Chr. 7 q34 | |
| T-cell receptor delta locus | |
|---|---|
| Identifiers | |
| Symbol | Chr. 14 q11.2 |
| T-cell receptor gamma locus | |
|---|---|
| Identifiers | |
| Symbol | Chr. 7 p14 |
The T-cell receptor (TCR) is a protein complex found on the surface of T cells, or T lymphocytes,[1] that is responsible for recognizing fragments of antigen as peptides bound to major histocompatibility complex (MHC) molecules. The binding between TCR and antigen peptides is of relatively low affinity and is degenerate: that is, many TCRs recognize the same antigen peptide and many antigen peptides are recognized by the same TCR.[2]
The TCR is composed of two different protein chains (that is, it is a heterodimer). In humans, in 95% of T cells the TCR consists of an alpha (α) chain and a beta (β) chain (encoded by TRA and TRB, respectively), whereas in 5% of T cells the TCR consists of gamma and delta (γ/δ) chains (encoded by TRG and TRD, respectively). This ratio changes during ontogeny and in diseased states (such as leukemia). It also differs between species. Orthologues of the 4 loci have been mapped in various species.[3][4] Each locus can produce a variety of polypeptides with constant and variable regions.[3]
When the TCR engages with antigenic peptide and MHC (peptide/MHC), the T lymphocyte is activated through
History
In 1982, Nobel laureate
Structural characteristics
The TCR is a disulfide-linked membrane-anchored heterodimeric protein normally consisting of the highly variable alpha (α) and beta (β) chains expressed as part of a complex with the invariant CD3 chain molecules. T cells expressing this receptor are referred to as α:β (or αβ) T cells, though a minority of T cells express an alternate receptor, formed by variable gamma (γ) and delta (δ) chains, referred as γδ T cells.[11]
Each chain is composed of two extracellular domains: Variable (V) region and a Constant (C) region, both of Immunoglobulin superfamily (IgSF) domain forming antiparallel β-sheets. The Constant region is proximal to the cell membrane, followed by a transmembrane region and a short cytoplasmic tail, while the Variable region binds to the peptide/MHC complex.
The variable domain of both the TCR α-chain and β-chain each have three hypervariable or complementarity-determining regions (CDRs). There is also an additional area of hypervariability on the β-chain (HV4) that does not normally contact antigen and, therefore, is not considered a CDR.[citation needed]
The residues in these variable domains are located in two regions of the TCR, at the interface of the α- and β-chains and in the β-chain
CDR2 is thought to recognize the MHC. HV4 of the β-chain is not thought to participate in antigen recognition as in classical CDRs, but has been shown to interact with superantigens.[13]
The constant domain of the TCR consists of short connecting sequences in which a cysteine residue forms disulfide bonds, which form a link between the two chains.
The TCR is a member of the immunoglobulin superfamily, a large group of proteins involved in binding, recognition, and adhesion; the family is named after
Generation of the TCR diversity
The generation of TCR diversity is similar to that for
Each recombined TCR possess unique antigen specificity, determined by the structure of the antigen-binding site formed by the α and β chains in case of αβ T cells or γ and δ chains on case of γδ T cells.[14]
- The TCR alpha chain is generated by VJ recombination, whereas the beta chain is generated by VDJ recombination (both involving a random joining of gene segments to generate the complete TCR chain).
- Likewise, generation of the TCR gamma chain involves VJ recombination, whereas generation of the TCR delta chain occurs by VDJ recombination.
The intersection of these specific regions (V and J for the alpha or gamma chain; V, D, and J for the beta or delta chain) corresponds to the CDR3 region that is important for peptide/MHC recognition (see above).
It is the unique combination of the segments at this region, along with
Later during development, individual CDR loops of TCR can be re-edited in the periphery outside thymus by reactivation of recombinases using a process termed TCR revision (editing) and change its antigenic specificity.
The TCR complex
In the plasma membrane the TCR receptor chains α and β associate with six additional adaptor proteins to form an octameric complex. The complex contains both α and β chains, forming the ligand-binding site, and the signaling modules
The signaling motifs involved in TCR signaling are tyrosine residues in the cytoplasmic tail of these adaptor proteins that can be phosphorylated in the event of TCR-pMHC binding. The tyrosine residues reside in a specific amino acid sequence of the signature Yxx(L/I)x6-8Yxx(L/I), where Y, L, I indicate tyrosine, leucine and isoleucine residues, x denotes any amino acids, the subscript 6-8 indicates a sequence of 6 to 8 amino acids in length. This motif is very common in activator receptors of the
Antigen discrimination
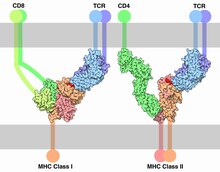
Each T cell expresses clonal TCRs which recognize a specific peptide loaded on a MHC molecule (pMHC), either on MHC class II on the surface of antigen-presenting cells or MHC class I on any other cell type.[16] A unique feature of T cells is their ability to discriminate between peptides derived from healthy, endogenous cells and peptides from foreign or abnormal (e.g. infected or cancerous) cells in the body.[17] Antigen-presenting cells do not discriminate between self and foreign peptides and typically express a large number of self-derived pMHCs on their cell surface and only a few copies of any foreign pMHC. For example, cells infected with HIV have only 8–46 HIV-specific pMHCs, compared with 100,000 total pMHCs, per cell.[18][19]
Because T cells undergo positive selection in the thymus, there is a non-negligible affinity between self-pMHC and the TCR. Nevertheless, the T-cell receptor signaling should not be activated by self-pMHC so that endogenous, healthy cells are ignored by T cells. However, when these very same cells contain even minute quantities of pathogen-derived pMHC, T cells must get activated and initiate immune responses. The ability of T cells to ignore healthy cells but respond when these same cells express a small number of foreign pMHCs is known as antigen discrimination.[20][21]
To do so, T cells have a very high degree of antigen specificity, despite the fact that the affinity to the peptide/MHC ligand is rather low in comparison to other receptor types.[22] The affinity, given as the dissociation constant (Kd), between a TCR and a pMHC was determined by surface plasmon resonance (SPR) to be in the range of 1–100 μM, with an association rate (kon) of 1000 -10000 M−1 s−1 and a dissociation rate (koff) of 0.01 -0.1 s−1.[23] In comparison, cytokines have an affinity of KD = 10–600 pM to their receptor.[24] It has been shown that even a single amino acid change in the presented peptide that affects the affinity of the pMHC to the TCR reduces the T-cell response and cannot be compensated by a higher pMHC concentration.[25] A negative correlation between the dissociation rate of the pMHC-TCR complex and the strength of the T-cell response has been observed.[26] That means, pMHC that bind the TCR for a longer time initiate a stronger activation of the T cell. Furthermore, T cells are highly sensitive; interaction with a single pMHC is enough to trigger activation.[27] T cells move on quickly from antigens that do not trigger responses, rapidly scanning pMHC on an antigen-presenting cell (APC) to increase the chance of finding a specific pMHC. On average, a T cell encounters 20 APCs per hour.[28]
Different models for the molecular mechanisms that underlie this highly specific and highly sensitive process of antigen discrimination have been proposed. The occupational model simply suggests that the TCR response is proportional to the number of pMHC bound to the receptor. Given this model, a shorter lifetime of a peptide can be compensated by higher concentration such that the maximum response of the T cell stays the same. However, this cannot be seen in experiments and the model has been widely rejected.[26] The most accepted view is that the TCR engages in kinetic proofreading. The kinetic proofreading model proposes that a signal is not directly produced upon binding but a series of intermediate steps ensure a time delay between binding and signal output. Such intermediate "proofreading" steps can be multiple rounds of tyrosine phosphorylation. These steps require energy and therefore do not happen spontaneously, only when the receptor is bound to its ligand. This way only ligands with high affinity that bind the TCR for a long enough time can initiate a signal. All intermediate steps are reversible, such that upon ligand dissociation the receptor reverts to its original unphosphorylated state before a new ligand binds.[29] This model predicts that maximum response of T cells decreases for pMHC with shorter lifetime. Experiments have confirmed this model.[26] However, the basic kinetic proofreading model has a trade-off between sensitivity and specificity. Increasing the number of proofreading steps increases the specificity but lowers the sensitivity of the receptor. The model is therefore not sufficient to explain the high sensitivity and specificity of TCRs that have been observed. (Altan Bonnet2005) Multiple models that extend the kinetic proofreading model have been proposed, but evidence for the models is still controversial.[17][30][31]
The antigen sensitivity is higher in antigen-experienced T cells than in naive T cells. Naive T cells pass through the process of functional avidity maturation with no change in affinity. It is based on the fact that effector and memory (antigen-experienced) T cell are less dependent on costimulatory signals and higher antigen concentration than naive T cell.[32]
Signaling pathway
The essential function of the TCR complex is to identify specific bound antigen derived from a potentially harmful pathogen and elicit a distinct and critical response. At the same time it has to ignore any self-antigen and tolerate harmless antigens such as food antigens. The signal transduction mechanism by which a T cell elicits this response upon contact with its unique antigen is termed T-cell activation. Upon binding to pMHC, the TCR initiates a signaling cascade, involving transcription factor activation and cytoskeletal remodeling resulting in T-cell activation. Active T cells secrete cytokines, undergo rapid proliferation, have cytotoxic activity and differentiate into effector and memory cells. When the TCR is triggered, T cells form an immunological synapse allowing them to stay in contact with the antigen presenting cell for several hours.[33] On a population level, T-cell activation depends on the strength of TCR stimulation, the dose–response curve of ligand to cytokine production is sigmoidal. However, T-cell activation on a single cell level can be characterized by a digital switch-like response, meaning the T cell is fully activated if the stimulus is higher than a given threshold; otherwise the T cell stays in its non-activated state. There is no intermediate activation state. The robust sigmoid dose-response curve on population level results from individual T cells having slightly different thresholds.[25]
T cells need three signals to become fully activated. Signal 1 is provided by the T-cell receptor when recognising a specific antigen on a MHC molecule. Signal 2 comes from co-stimulatory receptors on T cell such as CD28, triggered via ligands presented on the surface of other immune cells such as CD80 and CD86. These co-stimulatory receptors are expressed only when an infection or inflammatory stimulus is detected by the innate immune system, Known as a "Danger signal". This two-signal system makes sure that T cells only respond to harmful stimuli (i.e. pathogens or injury) and not to self-antigens. An additional third signal is provided by cytokines, which regulate the differentiation of T cells into different subsets of effector T cells.[33] There are a myriad of molecules involved in the complex biochemical process (called trans-membrane signaling) by which T-cell activation occurs. Below, the signaling cascade is described in detail.
Receptor activation
The initial triggering follows the mechanism common for all
Proximal TCR signaling
Phosphorylated ITAMs in the cytoplasmic tails of CD3 recruit protein tyrosine kinase
Molecules that bind the LAT/Slp76 complex include:
Signal transduction to the nucleus
PLCγ is a very important enzyme in the pathway as it generates second messenger molecules. It is activated by the tyrosine kinase Itk which is recruited to the cell membrane by binding to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 is produced by the action of phosphoinositide 3-kinase(PI-3K), which phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to produce PIP3. It is not known that PI-3K is activated by the T-cell receptor itself, but there is evidence that CD28, a co-stimulatory receptor providing the second signal, is able to activate PI-3K. The interaction between PLCγ, Itk and PI-3K could be the point in the pathway where the first and the second signal are integrated. Only if both signals are present, PLCγ is activated.[33] Once PLCγ is activated by phosphorylation. It hydrolyses PIP2 into two
These second messenger molecules amplify the TCR signal and distribute the prior localized activation to the entire cell and activate protein cascades that finally lead to the activation of transcription factors. Transcription factors involved in T-cell signaling pathway are the
NFAT
NFAT activation depends on calcium signaling. IP3 produced by PLC-γ is no longer bound to the membrane and diffuses rapidly in the cell. Binding of IP3 to calcium channel receptors on the endoplasmic reticulum (ER) induces the release of calcium (Ca2+) into the cytosol. The resulting low Ca2+ concentration in the ER causes STIM1 clustering on the ER membrane, which in turn leads to activation of cell membrane CRAC channels that allows additional calcium to flow into the cytosol from the extracellular space. Therefore, levels of Ca2+ are strongly increased in the T cell. This cytosolic calcium binds calmodulin, inducing a conformational change of the protein such that it can then bind and activate calcineurin. Calcineurin, in turn, dephosphorylates NFAT. In its deactivated state, NFAT cannot enter the nucleus as its nuclear localization sequence (NLS) cannot be recognized by nuclear transporters due to phosphorylation by GSK-3. When dephosphorylated by Calcineurin translocation of NFAT into the nucleus is possible.[33] Additionally, there is evidence that PI-3K via signal molecules recruits the protein kinase AKT to the cell membrane. AKT is able to deactivate GSK3 and thereby inhibiting the phosphorylation of NFAT, which could contribute to NFAT activation.[39]
NF-κB
NF-κB activation is initiated by DAG, the second, membrane bound product of PLCγ hydrolyzation of PIP2. DAG binds and recruits
AP1
Activation of AP1 involves three MAPK signaling pathways. These pathway use a phosphorylation cascade of three successive acting protein kinases to transmit a signal. The three MAPK pathways in T cells involve kinases of different specificities belonging to each of the MAP3K, MAP2K, MAPK families. Initial activation is done by the GTPase Ras or Rac which phosphorylate the MAP3K.[33] A cascade involving the enzymes
See also
References
- ISBN 978-1-4292-0211-4. Retrieved 28 November 2010.
- PMID 22918468.
- ^ PMID 11567625.
- PMID 16484802.
- ^ S2CID 1549902.
- S2CID 13249566.
- PMID 6185617.
- PMID 6601175.
- S2CID 4229210.
- S2CID 4273688.
- ^ Janeway Jr CA, Travers P, Walport M, et al. (2001). Immunobiology: The Immune System in Health and Disease. 5th edition. Glossary: Garland Science.
- PMID 10318939.
- PMID 17560120.
- ^ Janeway CA, Travers P, Walport M, et al. (2001). "The Generation of Lymphocyte Antigen Receptors". Immunobiology: The Immune System in Health and Disease (5th ed.). Garland Science.
- ^ PMID 12507424.
- PMID 19132916.
- ^ PMID 17825415.
- PMID 27401039.
- PMID 23298205.
- PMID 1833816.
- PMID 8879197.
- PMID 17082606.
- PMID 17442956.
- PMID 9789059.
- ^ PMID 16231973.
- ^ PMID 21653229.
- PMID 24120362.
- PMID 14722354.
- PMID 7761445.
- PMID 24636916.
- S2CID 14274400.
- PMID 22611418.
- ^ ISBN 978-0815345510.
- ^ S2CID 22423010.
- S2CID 44314604.
- PMID 20541955.
- PMID 11970992.
- S2CID 46343285.
- ^ PMID 19386893.
- ^ "UniProtKB - P06239 (LCK_HUMAN)". Uniprot. Retrieved 7 May 2020.
- PMID 9048554.
External links
- UMich Orientation of Proteins in Membranes protein/pdbid-2hac – Zeta-zeta dimer of T-cell receptor
- T-Cell+Receptor at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
| Authority control databases: National |
|---|