
Asymmetric hydrogenation
Asymmetric hydrogenation is a chemical reaction that adds two atoms of
History
In 1956 a
.svg)
The field of asymmetric hydrogenation continued to experience a number of notable advances.
Today, asymmetric hydrogenation is a routine methodology in laboratory and industrial scale organic chemistry. The importance of asymmetric hydrogenation was recognized by the 2001 Nobel Prize in Chemistry awarded to William Standish Knowles and Ryōji Noyori.
Mechanism
Asymmetric hydrogenations operate by conventional mechanisms invoked for other hydrogenations. This includes inner sphere mechanisms, outer sphere mechanisms and the σ-bond metathesis mechanisms.[7] The type of mechanism employed by a catalyst is largely dependent on the ligands used in a system, which in turn leads to certain catalyst-substrate affinities.
Inner sphere mechanisms
The so-called inner sphere mechanism entails coordination of the alkene to the metal center.[8] Other characteristics of this mechanism include a tendency for a homolytic splitting of dihydrogen when more electron-rich, low-valent metals are present while electron-poor, high valent metals normally exhibit a heterolytic cleavage of dihydrogen assisted by a base.[9]
The diagram below depicts purposed mechanisms for catalytic hydrogenation with rhodium complexes which are inner sphere mechanisms. In the unsaturated mechanism, the chiral product formed will have the opposite mode compared to the catalyst used. While the thermodynamically favoured complex between the catalyst and the substrate is unable to undergo hydrogenation, the unstable, unfavoured complex undergoes hydrogenation rapidly.[10] The dihydride mechanism on the other hand sees the complex initially hydrogenated to the dihydride form. This subsequently allows for the coordination of the double bond on the non-hindered side. Through insertion and reductive elimination, the product's chirality matches that of the ligand.[11]

The preference for producing one enantiomer instead of another in these reactions is often explained in terms of

Outer sphere mechanisms
Some catalysts operate by "outer sphere mechanisms" such that the substrate never bonds directly to the metal but rather interacts with its ligands, which is often a metal hydride and a protic hydrogen on a ligand. As such, in most cases dihydrogen is split heterolytically, with the metal acting as a Lewis acid and either an external or internal base "deprotonating" the hydride.[7]

For an example of this mechanism we can consider the BINAP-Ru-diamine system. The dihalide form of the catalyst is converted to the catalysts by reaction of H2 in the presence of base:[12]
- RuCl2(BINAP)(diamine) + 2 KOBu-t + 2 H2 → RuH2(BINAP)(diamine) + 2 KCl + 2 HOBu-t
The resulting catalysts have three kinds of ligands:
- hydrides, which transfer to the unsaturated substrate
- diamines, which interact with substrate and with base activator by the second coordination sphere
- diphosphine, which confers asymmetry.
The "Noyori-class" of catalysts are often referred to as bifunctional catalysts to emphasize the fact that both the metal and the (amine) ligand are functional.[13]
In the hydrogenation of C=O containing substrates, the mechanism was long assumed to operate by a six membered pericyclic transition state/intermediate whereby the hydrido ruthenium hydride center (HRu-NH) interacts with the carbonyl substrate R2C=O.[14] More recent DFT and experimental studies have shown that this model is largely incorrect. Instead, the amine backbone interacts strongly with the base activator, which often is used in large excess.[12] However in both cases, the substrate does not bond directly with the metal centre, thus making it a great example of an outer sphere mechanism.
Metals
Practical AH employ platinum metal-based catalysts.[15][16][17]
Base metals
Ligand classes
Phosphine ligands
Chiral
Chiral phosphine ligands can be generally classified as mono- or bidentate. They can be further classified according to the location of the stereogenic centre – phosphorus vs the organic substituents. Ligands with a C2 symmetry element have been particularly popular, in part because the presence of such an element reduces the possible binding conformations of a substrate to a metal-ligand complex dramatically (often resulting in exceptional enantioselectivity).[21]
Monodentate phosphines
Monophosphine-type ligands were among the first to appear in asymmetric hydrogenation, e.g., the ligand CAMP.
Chiral diphosphine ligands
The diphosphine ligands have received considerably more attention than the monophosphines and, perhaps as a consequence, have a much longer list of achievement. This class includes the first ligand to achieve high selectivity (DIOP), the first ligand to be used in industrial asymmetric synthesis (DIPAMP[27][28][4]) and what is likely the best known chiral ligand (BINAP).[3] Chiral diphosphine ligands are now ubiquitous in asymmetric hydrogenation.

P,N and P,O ligands


The use of P,N ligands in asymmetric hydrogenation can be traced to the C2 symmetric
NHC ligands
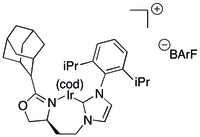
Simple
Some C,N ligands combine an NHC with a chiral oxazoline to give a chelating ligand.[35][36] NHC-based ligands of the first type have been generated as large libraries from the reaction of smaller libraries of individual NHCs and oxazolines.[35][36] NHC-based catalysts featuring a bulky seven-membered metallocycle on iridium have been applied to the catalytic hydrogenation of unfunctionalized olefins[35] and vinyl ether alcohols with conversions and ee's in the high 80s or 90s.[37] The same system has been applied to the synthesis of a number of aldol,[38] vicinal dimethyl[39] and deoxypolyketide[40] motifs, and to the deoxypolyketides themselves.[41]
C2-symmetric NHCs have shown themselves to be highly useful ligands for the asymmetric hydrogenation.[42]
Acyclic substrates
Substrates can be classified according to their polarity. Nonpolar substrates are dominated by
Nonpolar substrates
Alkenes that are particularly amenable to asymmetric hydrogenation often feature a polar functional group adjacent to the site to be hydrogenated. In the absence of this functional group, catalysis often results in low ee's. For some unfunctionalized olefins, iridium with P,N-based ligands) have proven effective, however. Alkene substrates are often classified according to their substituents, e.g., 1,1-disubstituted, 1,2-diaryl trisubstituted, 1,1,2-trialkyl and tetrasubstituted olefins.[44][45] and even within these classes variations may exist that make different solutions optimal.[46]


Conversely to the case of olefins, asymmetric hydrogenation of enamines has favoured diphosphine-type ligands; excellent results have been achieved with both iridium- and rhodium-based systems. However, even the best systems often suffer from low ee's and a lack of generality. Certain pyrrolidine-derived enamines of aromatic ketones are amenable to asymmetrically hydrogenation with cationic rhodium(I) phosphonite systems, and I2 and acetic acid system with ee values usually above 90% and potentially as high as 99.9%.[47] A similar system using iridium(I) and a very closely related phosphoramidite ligand is effective for the asymmetric hydrogenation of pyrrolidine-type enamines where the double bond was inside the ring: in other words, of dihydropyrroles.[48] In both cases, the enantioselectivity dropped substantially when the ring size was increased from five to six.
Imines and ketones

For carbonyl and imine substrates, end-on, η1 coordination can compete with η2 mode. For η1-bound substrates, the hydrogen-accepting carbon is removed from the catalyst and resists hydrogenation.[51]
Iridium/P,N ligand-based systems have been effective for some ketones and imines. For example, a consistent system for benzylic aryl imines uses the P,N ligand SIPHOX in conjunction with iridium(I) in a cationic complex to achieve asymmetric hydrogenation with ee >90%.[52] An efficient catalyst for ketones, (turnover number (TON) up to 4,550,000 and ee up to 99.9%) is an iridium(I) system with a closely related tridentate ligand.[53]

The BINAP/diamine-Ru catalyst is effective for the asymmetric reduction of both functionalized and simple ketones,

Aromatic substrates
The asymmetric hydrogenation of
Quinolines, isoquinolines and quinoxalines
Two systems exist for the asymmetric hydrogenation of 2-substituted
The second is an organocatalytic

Much of the asymmetric hydrogenation chemistry of quinoxalines is closely related to that of the structurally similar
Pyridines
The most-general method of asymmetric pyridine hydrogenation is actually a heterogeneous method, where asymmetry is generated from a chiral oxazolidinone bound to the C2 position of the pyridine. Hydrogenating such functionalized pyridines over a number of different heterogeneous metal catalysts gave the corresponding piperidine with the substituents at C3, C4, and C5 positions in an all-cis geometry, in high yield and excellent enantioselectivity. The oxazolidinone auxiliary is also conveniently cleaved under the hydrogenation conditions.[66]

Methods designed specifically for 2-substituted pyridine hydrogenation can involve asymmetric systems developed for related substrates like 2-substituted quinolines and quinoxalines. For example, an iridium(I)\chiral phosphine\I2 system is effective in the asymmetric hydrogenation of activated (alkylated) 2-pyridiniums[67] or certain cyclohexanone-fused pyridines.[68] Similarly, chiral Brønsted acid catalysis with a Hantzsh ester as a hydride source is effective for some 2-alkyl pyridines with additional activating substitution.[69]
Indoles and pyrroles
The asymmetric hydrogenation of indoles has been established with N-Boc protection.[70]

A Pd(TFA)2/H8-BINAP system achieves the enantioselective cis-hydrogenation of 2,3- and 2-substituted indoles.[71][72]

Akin to the behavior of indoles,

Oxygen- and sulfur-containing heterocycles
The asymmetric hydrogenation of furans and benzofurans is challenging.[74]

Asymmetric hydrogenation of

Heterogeneous catalysis
No heterogeneous catalyst has been commercialized for asymmetric hydrogenation.
The first asymmetric hydrogenation focused on palladium deposited on a silk support.

An alternative technique and one that allows more control over the structural and electronic properties of active catalytic sites is the immobilization of catalysts that have been developed for homogeneous catalysis on a heterogeneous support. Covalent bonding of the catalyst to a polymer or other solid support is perhaps most common, although immobilization of the catalyst may also be achieved by adsorption onto a surface, ion exchange, or even physical encapsulation. One drawback of this approach is the potential for the proximity of the support to change the behaviour of the catalyst, lowering the enantioselectivity of the reaction. To avoid this, the catalyst is often bound to the support by a long linker though cases are known where the proximity of the support can actually enhance the performance of the catalyst.[76]
The final approach involves the construction of MOFs that incorporate chiral reaction sites from a number of different components, potentially including chiral and achiral organic ligands, structural metal ions, catalytically active metal ions, and/or preassembled catalytically active organometallic cores.[77] One of these involved ruthenium-based catalysts. As little as 0.005 mol% of such catalysts proved sufficient to achieve the asymmetric hydrogenation of aryl ketones, although the usual conditions featured 0.1 mol % of catalyst and resulted in an enantiomeric excess of 90.6–99.2%.[78]

Industrial applications
-4_Ro_67-8867.svg)
Asymmetric hydrogenations are used in the production of several drugs, such as the antibacterial levofloxacin, the antibiotic carbapenem, and the antipsychotic agent BMS181100.[15][16][17]

Knowles' research into asymmetric hydrogenation and its application to the production scale synthesis of L-Dopa[4] gave asymmetric hydrogenation a strong start in the industrial world. A 2001 review indicated that asymmetric hydrogenation accounted for 50% of production scale, 90% of pilot scale, and 74% of bench scale catalytic, enantioselective processes in industry, with the caveat that asymmetric catalytic methods in general were not yet widely used.[79]
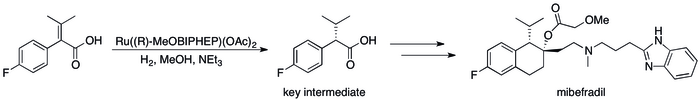
Asymmetric hydrogenation has replaced kinetic resolution based methods has resulted in substantial improvements in the process's efficiency.[12] can be seen in a number of specific cases where the For example, Roche's Catalysis Group was able to achieve the synthesis of (S,S)-Ro 67-8867 in 53% overall yield, a dramatic increase above the 3.5% that was achieved in the resolution based synthesis.[80] Roche's synthesis of mibefradil was likewise improved by replacing resolution with asymmetric hydrogenation, reducing the step count by three and increasing the yield of a key intermediate to 80% from the original 70%.[81]
Noyori-inspired hydrogenation catalysts have been applied to the commercial synthesis of number of fine chemicals. (R)-1,2-Propandiol, precursor to the antibacterial levofloxacin, can be efficiently synthesized from hydroxyacetone using Noyori asymmetric hydrogenation:[17]

Newer routes focus on the hydrogenation of (R)-methyl lactate.[12]
An antibiotic

An antipsychotic agent

References
- ^ "The Nobel Prize in Chemistry 2001". 2001-10-10.
- S2CID 4221816.
- ^ .
- ^ PMID 19746594.
- .
- PMID 15069193.
- ^ ISBN 978-3-527-31161-3.
- PMID 15379579.
- PMID 33480901.
- PMID 15379579.
- PMID 22192064.
- ^ S2CID 106394152.
- PMID 11722188.
- ^ PMID 17960897.
- ^ .
- ^ PMID 19746595
- PMID 18412184.
- PMID 19133772.
- PMID 22448656.
- .
- PMID 4270504.
- ^ .
- S2CID 95403641.
- PMID 12120551.
- PMID 12596201.
- .
- .
- .
- ^ PMID 10891051.
- PMID 29711115.
- PMID 21381140.
- PMID 22571628.
- PMID 21882320.
- ^ PMID 12515512.
- ^ PMID 16557626.
- .
- PMID 19368378.
- PMID 19719102.
- PMID 17200966.
- PMID 17338543.
- PMID 21442699.
- PMID 16159153.
- PMID 21140401.
- PMID 21556431.
- PMID 19658416.
- PMID 16953614.
- PMID 19132836.
- PMID 11169691.
- PMID 17576143.
- PMID 11722188.
- PMID 17002383.
- PMID 21751315.
- PMID 11169691
- ^ PMID 17896823.
- PMID 12940733.
- ^ PMID 15756313.
- hdl:10397/26878.
- PMID 17328554.
- PMID 16637664.
- PMID 16703140.
- PMID 16639754.
- PMID 19876991.
- PMID 20140920.
- PMID 15150766.
- PMID 22969060.
- hdl:10397/22884.
- PMID 17492817.
- PMID 20104554.
- PMID 21932274.
- PMID 21567504.
- PMID 21591641.
- PMID 22098109.
- PMID 22934527.
- ^ PMID 16802397.
- PMID 22084838.
- PMID 13129339.
- PMID 12613584.
- ISBN 978-3-527-63063-9.
- ISBN 978-3-540-64336-4.