
MicroRNA sequencing
MicroRNA sequencing (miRNA-seq), a type of
Introduction
History
MicroRNA sequencing (miRNA-seq) was developed to take advantage of
miRNA-seq can be performed using a variety of sequencing platforms. The first analysis of small
Methods
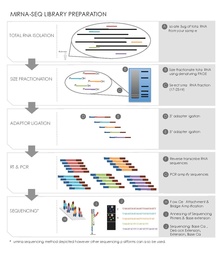
Small RNA Preparation
Sequence library construction can be performed using a variety of different kits depending on the high-throughput sequencing platform being employed. However, there are several common steps for small RNA sequencing preparation.[19][20]
Total RNA Isolation
In a given sample all the RNA is extracted and isolated using an isothiocyanate/phenol/chloroform (GITC/phenol) method or a commercial product such as Trizol (Invitrogen) reagent. A starting quantity of 50-100 μg total RNA, 1 g of tissue typically yields 1 mg of total RNA, is usually required for gel purification and size selection.[20] Quality control of the RNA is also measured, for example running an RNA chip on Caliper LabChipGX (Caliper Life Sciences).
Size Fractionation of small RNAs by Gel Electrophoresis
Isolated RNA is run on a denaturing polyacrylamide gel. An imaging method such as radioactive 5’-32P-labeled oligonucleotides along with a size ladder is used to identify a section of the gel containing RNA of the appropriate size, reducing the amount of material ultimately sequenced. This step does not have to be necessarily carried out before the ligation and reverse transcription steps outlined below.[19][20]
Ligation
The ligation step adds DNA adaptors to both ends of the small RNAs, which act as primer binding sites during reverse transcription and PCR amplification. An adenylated single strand DNA 3’adaptor followed by a 5’adaptor is ligated to the small RNAs using a ligating enzyme such as T4 RNA ligase2. The adaptors are also designed to capture small RNAs with a 5’ phosphate group, characteristic microRNAs, rather than RNA degradation products with a 5’ hydroxyl group.[19][20]
Reverse Transcription and PCR Amplification
This step converts the small adaptor ligated RNAs into cDNA clones used in the sequencing reaction. There are many commercial kits available that will carry out this step using some form of reverse transcriptase. PCR is then carried out to amplify the pool of cDNA sequences. Primers designed with unique nucleotide tags can also be used in this step to create ID tags in pooled library multiplex sequencing.[19][20]
Sequencing
The actual RNA sequencing varies significantly depending on the platform used. Three common next-generation sequencing
Data Analysis
Central to miRNA-seq data analysis is the ability to 1) obtain miRNA abundance levels from sequence reads, 2) discover novel miRNAs and then be able to 3) determine the differentially expressed miRNA and their 4) associated mRNA gene targets.
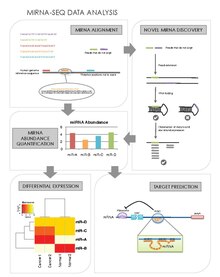
miRNA Alignment & Abundance Quantification
miRNAs may be preferentially expressed in certain cell types, tissues, stages of development, or in particular disease states such as cancer.
- After sequencing, the raw sequence reads are filtered based on quality. The adaptor sequences are also trimmed off the raw sequence reads.
- The resulting reads are then formatted into a fasta file where the copy number and sequence is recorded for each unique tag.
- Sequences that may represent E. Coli contamination are identified by a BLAST search against an E. Coli database and are removed from analysis.
- Each of the remaining sequences are aligned against a miRNA sequence database (such as miRBase[28]) In order to account for imperfect DICER processing, a 6nt overhang on the 3’ end, and 3nt on the 5’ end are allowed.
- The reads that do not align to the miRNA database are then loosely aligned to miRNA precursors to detect miRNAs that might carry mutations or those that have gone through RNA editing.
- The read counts for each miRNA are then normalized to the total number of mapped miRNAs to report the abundance of each miRNA.
Novel miRNA Discovery
Another advantage of miRNA-seq is that it allows the discovery of novel miRNAs that may have eluded traditional screening and profiling methods.[27] There are several novel miRNA discovery algorithms. Their general steps are as follows:
- Obtain reads that did not align to known miRNA sequences, and map them to the genome.
- RNA Folding Method
- For the miRNA sequences were an exact match is found, obtain the genomic sequence including ~100bp of flanking sequence on either side, and run the RNA through RNA folding software such as the Vienna package.[29]
- Folded sequences that lie on one arm of the miRNA hairpin and have a minimum free energy of less than ~25kcal/mol are shortlisted as putative miRNA.
- The shortlisted sequences are trimmed down to include only the possible precursor sequence and are then refolded to ensure that the precursor was not artificially stabilized by neighbouring sequences.
- The resulting folded sequences are considered novel miRNAs if the miRNA sequence falls within one arm of the hairpin, and are highly conserved between species.
- Star Strand Expression Method (miRdeep[30])
- Novel miRNA sequences are identified based on the characteristic expression pattern that they display due to DICER processing: higher expression of the mature miRNA over the star strand and loop sequences.
Differential Expression Analysis
After the abundances of miRNAs are quantified for each sample, their expression levels can be compared between samples. One would then be able to identify miRNA that are preferentially expressed that particular time points, or in particular tissues or disease states. After normalizing for the number of mapped reads between samples, one can use a host of statistical tests (like those used in gene expression profiling) to determine differential expression
Target Prediction
Identifying a miRNA's mRNA targets will provide an understanding of the genes or networks of genes whose expression they regulate.[31] Public databases provide predictions of miRNA targets. But to better distinguish true positive predictions from false positive predictions, miRNA-seq data can be integrated to mRNA-seq data to observe for miRNA:mRNA functional pairs. RNA22,[32] TargetScan,[33][34][35][36][37][38] miRanda,[39] and PicTar[40] are software designed for this purpose. A list of prediction software is given here. The general steps are:
- Determine miRNA:mRNA binding pairs, complementarity between the miRNA sequences at the 3’-UTR of the mRNA sequence is identified.
- Determine the degree of conservation of miRNA:mRNA binding pairs across species. Typically, more highly binding pairs are less likely to be false positives of prediction.
- Observe for evidence of miRNA targeting in mRNA-seq or protein expression data: where the miRNA expression is high, the gene and protein expression of its target gene should be low.
Target Validation for Cleaved mRNA Targets
Many miRNAs function to direct cleavage of their mRNA targets; this is particularly true in plants, and thus high-throughput sequencing methods have been developed to take advantage of this property of miRNAs by sequencing the uncapped 3' ends of cleaved or degraded mRNAs. These methods are known as
Applications
Identification of Novel miRNAs
miRNA-seq has revealed novel miRNAs that were previously eluded in traditional miRNA profiling methods. Examples of such findings are in embryonic stem cells,[25] chicken embryos,[43] acute lymphoblastic leukaemia,[44] diffuse large b-cell lymphoma and b-cells,[45] acute myeloid leukemia,[46] and lung cancer.[47]
Disease biomarkers
Micro RNAs are important regulators of almost all cellular processes such as survival, proliferation, and differentiation. Consequently, it is not unexpected that miRNAs are involved in various aspects of cancer through the regulation of onco- and tumor suppressor gene expression. In combination with the development of high-throughput profiling methods, miRNAs have been identified as biomarkers for cancer classification, response to therapy, and prognosis.[48] Additionally, because miRNAs regulate gene expression they can also reveal perturbations in important regulatory networks that may be driving a particular disorder.[48] Several applications of miRNAs as biomarkers and predictors of disease are given below.
| Cancer type | miRNAs α | Ref. |
|---|---|---|
| Breast | ||
| ER Status | miR-26a/b, miR-30 family, miR-29b, miR-155, miR-342, miR-206, miR-191 | [49][50][51][52] |
| PR status | let-7c, miR-29b, miR-26a, miR-30 family, miR-520g | [52][53] |
| HER2/neu status | miR-520d, miR-181c, miR-302c, miR-376b, miR-30e | [49][53] |
| Lung | ||
| Squamous vs non-squamous cell | miR-205 | [54] |
| Small cell vs non-small cell | miR-17-5p, miR-22, miR-24, miR-31 | [48] |
| Gastric | ||
| Diffuse vs intestinal | miR-29b/c, miR-30 family, miR-135a/b | [55] |
| Endometrial | ||
| Endometrioid vs uterine papillary | miR-19a/b, miR-30e-5p, miR-101, miR-452, miR-382, miR-15a, miR-29c | [56] |
| Renal | ||
| Clear cell vs papillary | miR-424, miR-203, miR-31, miR-126 | [57] |
| Oncocytoma vs chromophobe | miR-200c, miR-139-5p | [57] |
| Myeloma | ||
| with t(14;16) | miR-1, miR-133a | [58] |
| with t(4;14) | miR-203, miR-155, miR-375 | [58] |
| with t(11;14) | miR-125a, miR-650, miR-184 | [58] |
| Acute myeloid leukemia | ||
| with t(15;17) | miR-382, miR-134, miR-376a, miR-127, miR-299-5p, miR-323 | [59] |
| with t(8;21) or inv(16) | let-7b/c, miR-127 | [59] |
| with NPM1 mutations | miR-10a/b, let-7, miR-29, miR-204, miR-128a, miR-196a/b | [59][60] |
| with FLT3 ITD | miR-155 | [59][60][61] |
| Chronic lymphocytic leukemia | ||
| ZAP-70 levels and IgVH status | miR-15a, miR-195, miR-221, miR-155, miR-23b | [62] |
| Melanoma | ||
| with BRAF V600E | miR-193a, miR-338, miR-565 | [63] |
| Lymphoma | ||
| Diffuse Large B Cell Lymphoma | has-miR-128, has-miR-129-3p, has-miR-152, has-miR-155, has-miR-185, has-miR-193a-5p, has-miR-196b, has-miR-199b-3p, has-miR-20b, has-miR-23a, has-miR-27a, has-miR-28-5p, has-miR-301a, has-miR-331-3p, has-miR-365, has-miR-625, has-miR-9 | [45] |
αThis is not a comprehensive list of miRNAs involved with these malignancies.
Comparison With Other Methods of miRNA Profiling
The disadvantages of using miRNA-seq over other methods of miRNA profiling are that it is more expensive, generally requires a larger amount of total RNA, involves extensive amplification, and is more time-consuming than microarray and qPCR methods.[3] As well, miRNA-seq library preparation methods seem to have systematic preferential representation of the miRNA complement, and this prevents accurate determination of miRNA abundance.[64] At the same time, the approach is hybridization independent and therefore does not require a priori sequence information. Because of this, one can obtain sequences of novel miRNAs and miRNA isoforms (isoMirs), distinguish sequentially similar miRNAs, and identify point mutations.[65]
Platform Comparison of miRNA Profiling
| qPCR | Microarray | Sequencing | |
|---|---|---|---|
| Throughput time | ~6 hours | ~2 days | 1–2 weeks |
| Total RNA required | 500 ng | 100-1,000 ng | 500-5,000 ng |
| Dynamic range detected | Six orders of magnitude | Four orders of magnitude | Five or more orders of magnitude |
| Infrastructure and technical requirements | Few | Moderate | Substantial |
| Cost per sample (USD) | $400 | $250–$350 | $500–$700 |
References
- ^ PMID 21125669.
- PMID 22082764.
- ^ S2CID 6853222.
- S2CID 8360619.
- S2CID 2669459.
- ^ S2CID 86602746.
- S2CID 13103224.
- PMID 8252621.
- PMID 8252622.
- S2CID 7044929.
- S2CID 6391484.
- S2CID 4410288.
- S2CID 33480585.
- PMID 22144189.
- S2CID 1651848.
- S2CID 16838469.
- PMID 20459774.
- PMID 21716661.
- ^ PMID 17889797.
- ^ PMID 18158127.
- ^ S2CID 6384349.
- ^ "Applications - Transcriptome Sequencing : 454 Life Sciences, a Roche Company". Archived from the original on 2011-05-26. Retrieved 2012-03-01.
- ^ "Illumina DesignStudio".
- ^ "Archived copy". Archived from the original on 2008-05-16. Retrieved 2008-05-16.
{{cite web}}: CS1 maint: archived copy as title (link) - ^ PMID 18285502.
- PMID 18158128.
- ^ PMID 19332473.
- PMID 21037258.
- S2CID 19344304.
- PMID 21775303.
- PMID 22208850.
- S2CID 12749133.
- S2CID 17316349.
- PMID 17612493.
- PMID 18955434.
- PMID 21909094.
- PMID 26267216.
- PMID 30286781.
- PMID 17532529.
- ^ Krek, A. Identification of microRNA targets. DAI-B 70/07, (2010).
- S2CID 13187064.
- PMID 18472421.
- PMID 21171994.
- PMID 19724645.
- ^ PMID 20733160.
- PMID 20962326.
- PMID 22027949.
- ^ PMID 21354374.
- ^ PMID 17922911.
- PMID 18089790.
- PMID 16784538.
- ^ PMID 16103053.
- ^ PMID 19432961.
- PMID 19273703.
- PMID 20022810.
- PMID 20542546.
- ^ PMID 20595629.
- ^ PMID 20054351.
- ^ PMID 18337557.
- ^ PMID 18308931.
- PMID 18450603.
- PMID 16251535.
- PMID 20357817.
- S2CID 7953265.
- PMID 20360395.