
Bioconjugation
This article may be too technical for most readers to understand. (October 2022) |
Bioconjugation is a chemical strategy to form a stable covalent link between two molecules, at least one of which is a biomolecule.
Overview
Function
Recent advances in the understanding of biomolecules enabled their application to numerous fields like medicine, diagnostics, biocatalysis and materials. Synthetically modified biomolecules can have diverse functionalities, such as tracking cellular events, revealing
Types of Conjugated Molecules
The most common types of bioconjugation include coupling of a small molecule (such as
Protein-protein conjugations, such as the coupling of an antibody to an enzyme, or the linkage of protein complexes, is also facilitated via bioconjugations.[6][7]
Other less common molecules used in bioconjugation are
Common Bioconjugation Reactions
However, these reactions often lack
On Natural Amino Acids
Reactions of lysines
The

Reactions of cysteines
Because free cysteine rarely occurs on protein surface, it is an excellent choice for chemoselective modification.

Reactions of tyrosines
Tyrosine residues are relatively unreactive; therefore they have not been a popular targets for bioconjugation. Recent development has shown that the tyrosine can be modified through

Reactions of N- and C- termini
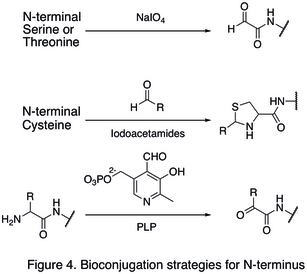
Since natural amino acid residues are usually present in large quantities, it is often difficult to modify one single site. Strategies targeting the termini of protein have been developed, because they greatly enhanced the site selectivity of protein modification. One of the N- termini modifications involves the functionalization of the terminal amino acid. The oxidation of N-terminal serine and threonine residues are able to generate N-terminal aldehyde, which can undergo further bioorthogonal reactions (shown in the first reaction in Figure 4). Another type of modification involves the condensation of N-terminal cysteine with aldehyde, generating thiazolidine that is stable at high pH (second reaction in Figure 4). Using pyridoxal phosphate (PLP), several N-terminal amino acids can undergo transamination to yield N-terminal aldehyde, such as glycine and aspartic acid (third reaction in Figure 4).
An example of C-termini modification is the native chemical ligation (NCL), which is the coupling between a C-terminal thioester and a N-terminal cysteine (Figure 5).

Bioorthogonal Reactions: On Unique Functional Groups
Modification of ketones and aldehydes
A ketone or aldehyde can be attached to a protein through the oxidation of N-terminal serine residues or transamination with PLP. Additionally, they can be introduced by incorporating

The introduction of nucleophilic catalyst can significantly enhance reaction rate (shown in Figure 7). For example, using

Recent developments that exploit proximal functional groups have enabled hydrazone condensations[21] to operate at 20 M−1s−1 at neutral pH while oxime condensations have been discovered which proceed at 500-10000 M−1s−1 at neutral pH without added catalysts.[22][23]
Staudinger ligation with azides
The

Contrasting with the classic Staudinger reaction, Staudinger ligation is a

Huisgen cyclization of azides
Copper catalyzed Huisgen cyclization of azides
Azide has become a popular target for chemoselective protein modification, because they are small in size and have a favorable thermodynamic reaction potential. One such azide reactions is the [3+2] cycloaddition reaction with alkyne, but the reaction requires high temperature and often gives mixtures of regioisomers.

An improved reaction developed by chemist

Strain promoted Huisgen cyclization of azides
Even though Staudinger ligation is a suitable bioconjugation in living cells without major toxicity, the phosphine's sensitivity to air oxidation and its poor

Transition Metal-Mediated Bioconjugation Reactions
Alkylation
On Natural Amino Acids
Using in situ generated RhII-carbenoid by activation of vinyl-substituted diazo compounds with Rh2(OAc)4, tryptophans and cysteines were shown to be selectively alkylated in aqueous media.
However, this method is limited to surface tryptophans and cysteines possibly because of steric constraints.[34]

- Ir-catalyzed Lys and N-terminus (reductive) alkylation[35]
Imines formed from the condensation of aldehydes with lysines or the N-terminus can be reduced efficient by an water-stable [Cp*Ir(bipy)(H2O)]SO4 complex in the presence of formate ions (serving as the hydride source). The reaction happens readily under physiologically relevant conditions and results in high conversion for various aromatic aldehydes.
_alkylation.png)
- Pd-catalyzed Tyr O-alkylation[36]
By using a pre-formed electrophilic π-allylpalladium(II) reagent derived from allylic acetate or carbamate precursors, selective allylic alkylation of tyrosines can be achieved in aqueous solution at room temperature and in the presence of cysteines.

- Au-catalyzed Cys alkylation[37]
Cysteine-containing peptides have been shown to undergo 1,2-addition to allenes in the presence of gold(I) and/or silver(I) salts, producing hydroxyl substituted vinyl thioethers. The reaction with peptides proceeds with high yields and is selective for cysteines over other nucleophilic residues.
However, the reactivity towards proteins is much decreased, potentially due to the coordination of gold to the protein backbone.

Arylation
On Natural Amino Acids
- Trp arylation
Multiple methods have been reported to achieve tryptophan C–H arylation, where diverse electrophiles such as aryl halides[38][39] and aryl boronic acids[40] (an example shown below) have been used to transfer the aryl groups.
However, current tryptophan C–H arylation reaction conditions remain relatively harsh, requiring organic solvents, low pH and/or high temperatures.

- Cys arylation
Free thiols has been considered unfavorable for Pd-mediated reactions due to Pd-catalyst decomposition.[41] However, PdII oxidative addition complexes (OACs) supported by dialkylbiaryl phosphine ligands have shown to work efficiently towards cysteine S-arylation.
The first example is the use of PdII OAC with
_-2.png)
There are other applications of this method where the PdII complexes were generated as PdII-peptide OACs by introducing 4-halophenylalanine into peptides during

Alternate to directly oxidative addition to the peptide, the Pd OACs could also be transferred to the protein through amine-selective acylation reaction via NHS ester. The latter has been applied to selectively label surface lysine residues of a protein (forming PdII-protein OACs) and oligonucleotides (forming PdII-oligonucleotide OACs), which could then be linked to cysteine-containing peptides or proteins.[46]

Another example of protein-protein cross-coupling is achieved through converting cysteine residues into an electrophilic S-aryl–Pd–X OAC by utilizing an intramolecular oxidative addition strategy.[47]
- Lys arylation[48]
Similar to cysteine, lysine N-arylation could be achieved through Pd OACs with different dialkylbiaryl phosphine ligands. Due to weaker nucleophilicity and slower reductive elimination rate compared to cysteine, the selection of supporting ligands is shown to be critical. The bulky BrettPhos and t-BuBrettPhos ligands in conjunction with mildly basic sodium phenoxide have been used as the strategy to functionalize lysines on peptide substrates. The reaction happens in mild conditions and is selective over most other nucleophilic amino acid residues.

On Unnatural Amino Acids
Pd-mediated

Examples of Applied Bioconjugation Techniques
Growth Factors
Bioconjugation of TGF-β to iron oxide nanoparticles and its activation through magnetic hyperthermia in-vitro has been reported.[58] This was done by using 1-(3-dimethylaminopropyl)ethylcarbodiimide combined with N-Hydroxysuccinimide to form primary amide bonds with the free primary amines on the growth factor. Carbon nanotubes have been successfully used in conjunction with bioconjugation to link TGF-β followed by an activation with near-infrared light.[59] Typically, these reactions have involved the use of a crosslinker, but some of these add molecular space between the compound of interest and base material and in turn causes higher degrees of non-specific binding and unwanted reactivity.[60]
See also
- Immunofluorescence
- Biomolecular engineering
- Biotinylation
- SpyTag/SpyCatcher
- In situ cyclization of proteins
- Unnatural amino acids
- Bioconjugate Chemistry journal
References
- ^ PMID 22086289.
- ^ ISBN 978-0470048672.
- ^ PMID 21112236.
- ^ PMID 20622973.
- PMID 20069754. Archived from the originalon February 2, 2014.
- PMID 26000775.
- PMID 38344167.
- S2CID 23657616.
- S2CID 137867074.
- ^ PMID 17450134.
- ^ PMID 15547999.
- PMID 11433435.
- ^ PMID 12203546.
- PMID 28443656.
- PMID 12034854.
- PMID 19229937.
- PMID 24410136.
- PMID 23534985.
- PMID 15584710.
- ^ PMID 17051631.
- PMID 24224646.
- PMID 25164607.
- PMID 29142692.
- PMID 12696879.
- PMID 18451302.
- S2CID 19720277.
- S2CID 4371934.
- PMID 15725026.
- PMID 20080615.
- S2CID 220272489.
- S2CID 103469980.
- PMID 15315433.
- PMID 23175246.
- ISSN 0002-7863.
- PMID 16190700.
- PMID 16433516.
- PMID 23322001.
- PMID 20013969.
- PMID 28074503.
- PMID 24516861.
- PMID 11483047.
- PMID 26511579.
- PMID 29081961.
- PMID 28777001.
- PMID 36320916.
- PMID 33730425.
- PMID 32364380.
- PMID 28206688.
- S2CID 19601358.
- PMID 23641876.
- PMID 21899368.
- PMID 25347611.
- PMID 19852502.
- PMID 21206952.
- PMID 22175226.
- PMID 23440916.
- PMID 18816552.
- PMID 31261853.
- PMID 25775150.
- S2CID 103064199.