
Enolate
In
Bonding and structure

Enolate anions are electronically related to allyl anions. The anionic charge is delocalized over the oxygen and the two carbon sites. Thus they have the character of both an alkoxide and a carbanion.[5]
Although they are often drawn as being simple salts, in fact they adopt complicated structures often featuring aggregates.[6]
%3DCMe2(Litmeda)_dimer_(VITLUG).png)
Preparation
Deprotonation of enolizable ketones, aromatic alcohols, aldehydes, and esters gives enolates.[8][9] With strong bases, the deprotonation is quantitative. Typically enolates are generated from using lithium diisopropylamide (LDA).[10]
Often, as in conventional Claisen condensations, Mannich reactions, and aldol condensations, enolates are generated in low concentrations with alkoxide bases. Under such conditions, they exist in low concentrations, but they still undergo reactions with electrophiles. Many factors affect the behavior of enolates, especially the solvent, additives (e.g. diamines), and the countercation (Li+ vs Na+, etc.). For unsymmetrical ketones, methods exist to control the regiochemistry of the deprotonation.[11]

The deprotonation of carbon acids can proceed with either
Enolates can be trapped by

Role of Lewis acids on enolate formation
In addition to the use of strong bases, enolates can be generated using a

For

Geometry of enolates
Extensive studies have been performed on the formation of enolates. It is possible to generate, in most cases, the desired enolate geometry:[14]

For ketones, most enolization conditions give Z enolates. For esters, most enolization conditions give E enolates. The addition of HMPA is known to reverse the stereoselectivity of deprotonation.

The stereoselective formation of enolates has been rationalized with the Ireland model,[15][16][17][18] although its validity is somewhat questionable. In most cases, it is not known which, if any, intermediates are monomeric or oligomeric in nature; nonetheless, the Ireland model remains a useful tool for understanding enolates.

In the Ireland model, the deprotonation is assumed to proceed by a six-membered or cyclic[19] monomeric transition state. The larger of the two substituents on the electrophile (in the case above, methyl is larger than proton) adopts an equatorial disposition in the favored transition state, leading to a preference for E enolates. The model clearly fails in many cases; for example, if the solvent mixture is changed from THF to 23% HMPA-THF (as seen above), the enolate geometry is reversed, which is inconsistent with this model and its cyclic transition state.
Regiochemistry of enolate formation
If an unsymmetrical ketone is subjected to base, it has the potential to form two regioisomeric enolates (ignoring enolate geometry). For example:

The trisubstituted enolate is considered the
In general, kinetic enolates are favored by cold temperatures, conditions that give relatively ionic metal–oxygen bonding, and rapid deprotonation using a slight excess of a strong, sterically hindered base. The large base only deprotonates the more accessible hydrogen, and the low temperatures and excess base help avoid equilibration to the more stable alternate enolate after initial enolate formation. Thermodynamic enolates are favored by longer equilibration times at higher temperatures, conditions that give relatively covalent metal–oxygen bonding, and use of a slight sub-stoichiometric amount of strong base. By using insufficient base to deprotonate all of the carbonyl molecules, the enolates and carbonyls can exchange protons with each other and equilibrate to their more stable isomer. Using various metals and solvents can provide control over the amount of ionic character in the metal–oxygen bond.
Reactions
As powerful nucleophiles, enolates react readily with a variety of electrophiles. These reactions generate new C-C bonds and often new stereocenters. The stereoselectivity and regioselectivity is influenced by additives, solvent, counterions, etc. One important class of electrophiles are alkyl halides, and in this case a classic problem arises: O-alkylation vs C-alkylation. Controlling this selectivity has drawn much attention. The negative charge in enolates is concentrated on the oxygen, but that center is also highly solvated, which leads to C-alkylation.[20]
Other important electrophiles are aldehydes/ketones and

Sample aldol reaction with lithium enolate

Synthesis of enones using regiospecific enolate formation and masked functionality


Regiospecific formation is the controlled enolate formation by the specific deprotonation at one of the α-carbons of the ketone starting molecule. This provides one of the best understood synthetic strategies to introduce chemical complexity in
When
An enone can also serve as a precursor for regiospecific formation of an enolate, here the enone is a "masked functionality" for the enolate. This process is first described by Gilbert Stork[22] who is best known for his contributions to the study of selective enolate formation methods in organic synthesis. Reacting an enone with lithium metal generates the enolate at the α-carbon of the enone. The enolate product can either be trapped or alkylated. By using "masked functionality", it is possible to produce enolates that are not accessible by traditional methods.
The "masked functionality" approach to regiospecific enolate formation has been widely used in the total synthesis of natural products. For example, in the total synthesis of the steroid hormone progesterone,[23] Stork and co-workers used the "masked functionality" to stereospecifically construct one of the quaternary carbons in the molecule.
Aza enolates
Aza enolates (also known as imine anions, enamides, metallated Schiff bases, and metalloenamines) are nitrogen analogous to enolates.[24] When imines get treated with strong bases such as LDA, highly nucleophilic aza enolates are generated.
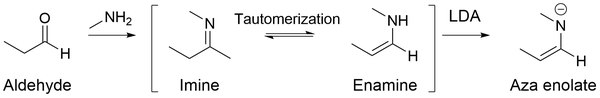
The major benefit of using aza enolates is that they don't undergo
On the other hand, imine has less electronegative nitrogen which induces a weaker partially positive charge on the carbonyl-carbon. As a result, while imines can still react with organolithiums, they don't react with other nucleophiles (including aza enolates) to undergo nucleophilic additions.[26]
Instead, aza enolates react similarly to enolates, forming
Two potential reaction mechanisms are shown below:

Since epoxide is a three-membered ring molecule, it has a high degree of ring strain. Although the carbons in the ring system are tetrahedral, preferring 109.5 degrees between each atom, epoxide strains the ring angles into 60 degrees. To counter this effect, the nucleophilic aza enolates easily react with epoxides to reduce their ring strains.

Besides reacting with epoxides, aza enolates can also react with
Aza enolates can also be formed with
See also
References
- ISBN 978-0-470-68253-1.
- .
- PMID 16836294.
- PMID 32040305.
- PMID 23941648.
- S2CID 97183362.
- ISBN 978-0-471-72091-1
- ISBN 978-3-527-67106-9.
- .
- .
- .
- ISBN 0-471-26418-0.
- .
- .
- .
- .
- PMID 11671880.
- ^ Directed Aldol Synthesis – Formation of E-enolate and Z-enolate
- ISBN 978-0-471-72091-1
- .
- ^ Stork, G.; Singh, J., J. Am. Chem. Soc. 1974, 96, 6181.
- ^ Stork, G.; McMurry, J. E., J. Am. Chem. Soc. 1967, 89, 5464.
- ^ a b c Aslam, O. (28 September 2012). Development of catalytic aza enolate reactions (Doctoral). UCL (University College London).
- ^ ISBN 978-0-19-927029-3.
- ^ Cranwell, Philippa. "Enamines/aza-enolates – Mechanism Mordor". sites.google.com. Archived from the original on 2021-09-03. Retrieved 2020-11-28.
- ISBN 978-0-387-68350-8.
- .
- ^ .
| Authority control databases: National |
|---|