
User:Lefh6/sandbox

Peroxisome proliferator-activated receptor agonists or PPAR agonists, are a group of drugs that induce the peroxisome proliferator-activated receptors (PPARs). The PPARs are a group of receptors found in the cell nucleus that act as important transcription factors for a variety of genes [1]. These genes are generally considered important in cell differentiation as well as in metabolic processes such as lipid and glucose homeostasis [1].
The

Thiazolidinediones (TZDs) are the only group of PPAR-γ agonists that have been marketed for type-2 diabetes mellitus. Two, rosiglitazone and pioglitazone are currently available in the United States, however only pioglitazone is available in Europe. Approval of the European Medicines Agency (EMA) for rosiglitazone was withdrawn in 2010 due to cardiovascular safety concerns [1].
The PPARs

The peroxisome proliferator-activated receptor agonists (PPARs) are a group of hormone receptors located in the cell nucleus that act as important, ligand activated transcription factors for a variety of genes. The PPARs perform various activities, mainly through endogenous ligands, in the metabolism of fatty acids as well by influencing energy balance through glucose regulation [1]. The PPAR family consists of three isoforms PPAR-α, PPAR-β/δ and PPAR-γ that differ from each other in ligand specificity, physiological roles and tissue distribution. All three isoforms contribute to expression of the gene systems that are involved in lipid metabolism, inflammation, and metabolic homeostatis [1] [6]. The PPARs natural agonists consist of lipophilic substances, mainly fatty acids and their metabolites [6].
PPAR-α are expressed in numerous tissues in rodents and humans, especially in tissues with high metabolic activity and capacity for fatty acid oxidation, liver, kidney, heart, skeletal muscle, intestinal mucosa and brown fat [3] [7]. PPAR-α have an important role in fatty acid oxidation in the liver. PPAR-α agonists have therefore been indicated for triglyceride lowering effects related to decreased synthesis and increased catabolism of VLDL as well as increased HDL concentrations [1] [8]. Fibrates are a class of PPAR-α agonists that have been marketed for their effects on arteriosclerosis [1] [9].
PPAR-β/δ is expressed throughout the body but in relatively high levels in the brain and skin as well as adipose tissue [3] [6]. PPAR-β/δ take part in the control of lipid and cholesterol metabolism and apparently control fat consumption in animals as well as contributing to glucose regulation. No PPAR-β/δ agonists have been marketed, however they are believed to have a role in the treatment of obesity [1].
PPAR-γ are expressed in nearly all cells, but mainly in adipose tissue and to a lesser extent in the heart, intestine, spleen, skeletal muscle, liver and macrophages [1] [3]. The PPAR-γ gene contains of three promoters, PPAR-γ1, PPAR-γ2 and PPAR-γ3, that give different receptor isoforms. The PPAR-γ1 and PPAR-γ3 promoter sequences translate into the identical PPAR-γ1 protein while the PPAR-γ2 promoter is translated into a different protein isoform, PPAR-γ2. The PPAR-γ1 isoform is present in almost all cells, but the PPAR-γ2 isoform is mostly limited to adipose tissue. PPAR-γ2 is considered a more potent receptor than PPAR-γ1 [1] [3]. PPARγ receptor influences the macronutrient metabolism and is a target for thiazolidinediones which include synthetic insulin sensitizers. Furthermore, this receptor is essential in adipogenesis process by which it controls lipid metabolism and activation seems to limit inflammation [1] [5] [10].
The mechanism of action of PPAR-γ agonists
PPAR-γ agonists generally improve insulin and glucose parameters and increase insulin sensitivity [1]. The agonists mediate their effects mainly in adipose tissue. They influence the expression of genes involved in lipid uptake, -metabolism and insulin action resulting in enhanced insulin signaling, lipid uptake and anabolic lipid metabolism. At the same time they diminish lipolysis and free fatty acid (FFA) release [5]. This causes lipid concentrations in adipose tissue to rise and FFA concentration is minimized [1] [5]. The hypothesis is, that by repartitioning the lipids away from the two primary tissues responsible for insulin-related glucose metabolism, liver and muscle, PPAR-γ agonists improve hyperglycemia and reverse lipotoxicity induced insulin resistance [1] [5].
The PPAR ligand binding site
The PPARs are hormone receptors and have a
The LBD structure is mostly common between the three PPAR isoforms, with only minor differences. The PPAR-β/δ binding site is generally smaller than that of the other subtypes (PPAR-α and PPAR-γ) which could be the reason for the greater ligand specificity of that subtype. The PPAR-α has a more lipophilic LBD than the two other subtypes which causes it to be able to bind more lipophilic saturated fatty acids. It is however unable to bind certain potent PPAR-γ ligands [10].
Ligand binding stabilizes the LBD structure
Structure activity relationship (SAR)

A typical PPAR-γ agonist consists of an acidic head, a central aromatic scaffold and a hetero-aromatic hydrophobic tail [2] [12] [13]. Flexible linkers connect the three pharmacophore centers. These linkers adapt to the curved binding site and are sometimes branched to access additional subpockets [14].
Lipophilicity has proved to be an important indicator of PPAR agonist binding affinity as well as molecular size.The acidic head is generally involved in the formation of a hydrogen bond network upon receptor binding. The acidic head is essential to the structure as research has shown that compounds without an acidic head are inactive [12] [13]. The acidic head also contributes to salt bridge formation with a positively charged receptor site. The PPAR-γ class of agonists is subdivided into five chemical groups, thiazolidinediones, tyrosine-based, indole-based, propionic-acid and phenylacetic acid derivatives. The groups show progressive differences in binding affinities. [2].
Thiazolidinedione analogs
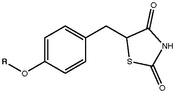
TZDs posess a 5-benzyl-1,3-thiazolidine-2,4-dione moiety that is neccessary for binding to the PPAR-γ. However research has shown that compounds containing a pyridine ring attached to the thiazolidine-2,4-dione ring system instead of benzene can also bind to the PPAR-γ [15]. The thiazolidinedione ring system contains three hydrogen bond acceptors but only one hydrogen bond donor, the NH group. These groups are responsible for the formation of the neccessary hydrogen bond network [15].
The oxygen atom connected to the aromatic ring in the form of an ether has proved to be essential for the activity of TZD compounds. A two carbon atom linkage between the ether oxygen and terminal hydrophobic tail is optimal for activity. An extension of the carbon linkage chain may however decrease activity [15]. Substitution of the carbon atoms of the linker can lead to decreased activity as well as replacing those carbon atoms with electronegative groups has the same effect [15]. Steric bulk of the hydrophobic ring system is considered favorable. Substituting the ring at the point of connection or at the ortho position leads to increased activity [15].
Tyrosine analogs
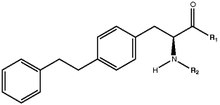
Tyrosine analogs contribute to the largest subclass of PPAR-γ agonists, generating interest because of the toxic side effects reported for TZDs [2]. A strong activity has been attributed to tyrosine analogs due to their ability to form a greater number of hydrogen bonds [2]. Tyrosine based compounds generally have two hydrogen bond donor sites (the carboxylic OH group and the NH group) and therefore show a greater hydrogen bonding affinity than TZD analogs that only contain one hydrogen bond donor site [2].
Bulk substitution of the tyrosine nitrogen atom of tyrosine analogs seems to contribute to a higher affinity with an area of the ligand binding domain [2]. The bridge oxygen, as well as the electronic cloud over the bridge and a carboxylic carbonyl group have been identified as essential parts of the tyrosine analog pharmacophore [2]. These groups are electron rich sites that may be involved in the formation of electrostatic, ionic, p-p and hydrogen bonds with the receptor [2].
Indole analogs

For indole derivatives, lipophilicity has proved to be the most important indicator of binding affinity followed by molecular weight. The indole ring itself has a strong lipophilic interactoin with PPAR- γ. Furthermore the derivatives can show a strong hydrophobic interaction promoted by a hydrophobic tail [2] [12] [13]. Furthermore flexibility and non-polar surfaces seem to exert a negative effect on the binding affinity of indole analogs. However, electrophilicity is not essential for tyrosine analogs [2]. A C5 substituted indole analog shows a better binding affinity, as well as functional activity, than C4 and C6 substituted analogs [12]. An increased distance from the carboxylic acid head to the indole nitrogen seems to decrease activity. Ethyl substitution on the carboxylic acid head seems to limit activity [12]. Thus research indicates that variation in linker length, the position on the indole and the tail building blocks seem to influence the agonists specificity for the PPAR subtypes [12].
History and development
In the 1950s the first fibrates (PPAR-α agonists) were synthesized as a result of a series of experiments conducted in France. Around 80 synthesized structures, derived from phenylethyl acetic acid,
.The first PPAR-γ agonists, thiazolidinediones were not discovered until late 20th century. Their discovery came as a surprise when Japanese researchers attempted to develop more
The discovery of ciglitazone was followed by the development of
In parallel, Takeda Laboratories developed pioglitazone that had similar effect in patients with type-2 diabetes mellitus [24] [8] Rosiglitazone and pioglitazone were marketed in the United states in 1999 for the treatment of type-2 diabetes mellitus. They are currently the only PPAR-γ agonists available in the USA. Only pioglitazone is available in Europe. Approval of the EMA for rosiglitazone was withdrawn in 2010 due to cardiovascular safety concerns [2] [8].
Current status and future perspectives
The thiazolidinediones class of drugs has exhibited unpleasant and dangerous side effects including, weight gain, edema, bone fractures and heart failure and increased risk of myocardial infractions. The reversible congestive heart failure that can occur has been linked to the body's inability to tolerate fluid retention that these drugs can cause. New partial PPAR-γ agonists are in development that should cause minimal side effects compaired to the more potent full agonists available today [1] [10].
TZDs have been shown to effectively lower glucose concentration and increase insulin sensitivity [4]. Diabetic patients do in general suffer from both hyperglycemia and dyslipidemia, and diabetes increases the risk of the development of arteriosclerosis [5]. As PPAR-γ agonists decrease the inflammation of adipose tissue that is induced by obesity, they have sparked interest for a possible role in limiting arterioclerosis as well as treating diabetes [10]. However the TZDs effect on blood pressure and lipid metabolism has been a disappointment. These drugs have not been able to reduce the need to treat dyslipidemia and hypertension with additional therapies [4].
This has lead to research of dual PPARα and PPARγ agonists, glitazars, in the hope of developing drugs with broad beneficial metabolic effects through simultaneous treatment of hyperglycemia and dyslipidemia [1] [10]. However recent studies have shown the increased cardiovascular risk that has been contributed to current PPAR-γ agonists as well as showing increased risk of bladder cancer and renal dysfunction [1] [10].
See also
References
- ^ PMID 24524207.)
{{cite journal}}: CS1 maint: unflagged free DOI (link - ^ PMID 19689404.
- ^ PMID 15629253.
- ^ PMID 15356308.
- ^ PMID 15860371.
- ^ S2CID 23791899.
- ^ PMID 23119024.)
{{cite journal}}: CS1 maint: unflagged free DOI (link - ^ PMID 20393155.
- PMID 15142970.
- ^ S2CID 2240461.
- ^ PMID 17317294.
- ^ PMID 16366601.
- ^ PMID 16451087.
- PMID 16737814.
- ^ S2CID 44036278.
- S2CID 30905313.
- S2CID 4306126.
- S2CID 3148132.
- PMID 7190396.
- ^ S2CID 43310974.
- PMID 9518284.
- PMID 7966158.
- S2CID 21527540.
- PMID 2339998.