
Autosomal dominant polycystic kidney disease
| Autosomal dominant polycystic kidney disease | |
|---|---|
| Other names | Autosomal dominant PKD, adult-onset PKD |
 | |
| Polycystic kidneys | |
| Specialty | Medical genetics |
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common, life-threatening
Signs and symptoms
Among the clinical presentation are:[citation needed]
- Acute loin pain
- Blood in the urine
- Ballotable kidneys
- Subarachnoid hemorrhage (berry aneurysm)
- Hypertension
- Associated liver cysts
- Uremia due to kidney failure
- Anemia due to chronic kidney disease
- Increase RBC or erythropoietin secretion
Signs and symptoms of ADPKD often develop between 30 and 40 years of age.[11]
Genetics
ADPKD is genetically heterogeneous with two
Large interfamilial and intrafamilial variability occurs in ADPKD.[1] Most individuals with PKD1 mutations have kidney failure by age 70 years, whereas more than 50% of individuals with PKD2 mutations have adequate renal function at that age (mean age of onset of end-stage renal disease: 54·3 years with PKD1; 74·0 years with PKD2).[19]
The significant intrafamilial variability observed in the severity of renal and extrarenal manifestations points to genetic and environmental modifying factors that may influence the outcome of ADPKD, and results of an analysis of the variability in renal function between monozygotic twins and siblings support the role of
Pathophysiology
In many patients with ADPKD, kidney dysfunction is not clinically apparent until 30 or 40 years of life.[5] However, an increasing body of evidence suggests the formation of renal cysts starts in utero.[24] Cysts initially form as small dilations in renal tubules, which then expand to form fluid-filled cavities of different sizes.[24] Factors suggested to lead to cystogenesis include a germline mutation in one of the polycystin gene alleles, a somatic second hit that leads to the loss of the normal allele, and a third hit, which can be a renal insult that triggers cell proliferation, and an injury response.[25] Due to numerous similarities between the pathophysiology of ADPKD and the pathophysiology of the renal response to injury, ADPKD has been described as a state of aberrant and persistent activation of renal injury response pathways.[26] In the progression of the disease, continued dilation of the tubules through increased cell proliferation, fluid secretion, and separation from the parental tubule lead to the formation of cysts.[27][28]
ADPKD, together with many other diseases that present with renal cysts, can be classified into a family of diseases known as
Epithelial cell proliferation and fluid secretion that lead to cystogenesis are two hallmark features in ADPKD.
Clinically, the insidious increase in the number and size of renal cysts translates as a progressive increment in kidney volume.[1][24] Studies led by Mayo Clinic professionals established that the total kidney volume (TKV) in a large cohort of ADPKD patients was 1060 ± 642ml with a mean increase of 204ml over three years, or 5.27% per year in the natural course of the disease, among other important, novel findings that were extensively studied for the first time.[33]

Diagnosis
Usually, the diagnosis of ADPKD is initially performed by renal imaging using
The findings of large
Molecular
-
 Adult polycystic kidney
Adult polycystic kidney -
 Diagram of autosomal dominant polycystic disease with a normal kidney inset for comparison
Diagram of autosomal dominant polycystic disease with a normal kidney inset for comparison -
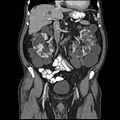 Abdominal CT scan of an adult with autosomal dominant polycystic kidney disease: Extensive cyst formation is seen over both kidneys, with a few cysts in the liver, as well. (Coronal plane)
Abdominal CT scan of an adult with autosomal dominant polycystic kidney disease: Extensive cyst formation is seen over both kidneys, with a few cysts in the liver, as well. (Coronal plane)



Treatment
Currently, the only pharmacological treatment available for ADPKD consists in reducing the speed in gain of total kidney volume (TKV) with vasopressin receptor 2 (V2) antagonists (i.e. tolvaptan).[38] Tolvaptan treatment does not halt or reverse disease progression and patients still progress towards renal failure. Palliative treatment modalities involve symptomatic medications (nonopioid and opioid analgesics) for abdominal/retroperitoneal pain. Options for analgesic-resistant pain include simple or complex surgical procedures (i.e. renal cyst aspiration, cyst decortication, renal denervation and nephrectomy), which can result in complications inherent to surgery.[citation needed] Recent research suggests that ketogenic dietary interventions beneficially affect the progression and symptoms in individuals with ADPKD.[39] Mild weight loss favorably affects pain[40] indicating the benefit of dietary and lifestyle changes.
Aquaretic medication
In 2014, Japan was the first country in the world to approve a pharmacological treatment for ADPKD
Dietary and lifestyle interventions
Research using ADPKD mouse models showed that mild food restriction strongly improved disease progression.
Dietary intake of sodium is associated with worse renal function decline in ADPKD,[55] and limiting sodium intake is generally recommended to patients. Dietary protein intake was not found to correlate with ADPKD progression.[56]
Increased water intake is thought to be beneficial in ADPKD and is generally recommended.[57][58] The underlying beneficial mechanism of increased water intake may be related to effects on the vasopressin V2 receptor or may be due to the suppression of harmful micro-crystal formation in renal tubules by dilution of solutes such as calcium oxalate, calcium phosphate and uric acid.[57][59]
Dietary intake of oxalate or inorganic phosphate has been shown to accelerate PKD disease progression in several rodent models.[57] Low levels or urinary citrate – a natural antagonist of the formation of harmful crystals in kidney tubules – have been shown to associate with worse disease progression in ADPKD patients.[57]
Analgesic medication
Renal cyst aspiration
Aspiration with ethanol sclerotherapy can be performed for the treatment of symptomatic simple renal cysts, but can be impractical in advanced patients with multiple cysts.[61] The procedure itself consists in the percutaneous insertion of a needle into the identified cyst, under ultrasound guidance, with subsequent draining the contained liquid; the sclerotherapy is used to avoid liquid reaccumulation that can occur in the cyst, which can result in symptom recurrence.[61][62]
Laparoscopic cyst decortication
Laparoscopic cyst decortication (also referred to as marsupialization) consists in the removal of one or more kidney cysts through
Neurolysis
A novel treatment of specifically the chronic pain experienced by many with ADPKD is
Nephrectomy
Many ADPKD patients experience symptomatic sequelae in consequence of the disease, such as cyst
Dialysis
Two modalities of dialysis can be used in the treatment of ADPKD patients: peritoneal dialysis and hemodialysis.[75] Epidemiological data shows that ADPKD affects 5–13.4% of patients undergoing hemodialysis in Europe and in the United States,[76][77][78] and about 3% in Japan.[7] Peritoneal dialysis has usually been contra-indicated in ADPKD patients with large kidney and liver volumes, due to expected physical difficulties in the procedure and possible complications;[75][79] however, no difference is seen in long-term morbidity between hemodialysis and peritoneal dialysis in ADPKD.[75]
Kidney transplant
Kidney transplantation is accepted as the preferred treatment for ADPKD patients with ESRD.[1] Among American patients on the kidney-transplant waiting list (as of December 2011), 7256 (8.4%) were listed due to cystic kidney disease and of the 16,055 renal transplants performed in 2011, 2057 (12.8%) were done for patients with cystic kidney disease, with 1,189 from deceased donors and 868 from living donors.[80]
Prognosis
In ADPKD patients, gradual cyst development and expansion result in kidney enlargement, and during the course of the disease,
Besides TKV and HtTKV, the estimated glomerular filtration rate (eGFR) has also been tentatively used to predict the progression of ADPKD.[81] After the analysis of CT or MRI scans of 590 patients with ADPKD treated at the Mayo Translational Polycystic Kidney Disease Center, Irazabal and colleagues developed an imaging-based classification system to predict the rate of eGFR decline in patients with ADPKD.[81][44] In this prognostic method, patients are divided into five subclasses of estimated kidney growth rates according to age-specific HtTKV ranges (1A, <1.5%; 1B, 1.5–3.0%; 1C, 3.0–4.5%; 1D, 4.5–6.0%; and 1E, >6.0%) as delineated in the CRISP study.[81][44] The decline in eGFR over the years following initial TKV measurement is significantly different between all five patient subclasses, with those in subclass 1E having the most rapid decline.[81] Some of the most common causes of death in patients with ADPKD are various infections (25%), a ruptured berry aneurysm (15%), or coronary/hypertensive heart disease (40%).[82]
References
- ^ S2CID 1700992.
- ^ "What is ADPKD?". PKD Foundation. Retrieved 2022-09-23.
- PMID 13469269.
- PMID 19455193.
- ^ PMID 20009161.
- S2CID 12124902.
- ^ S2CID 22124996.
- PMID 10972657.
- ^ PMID 23121377.
- ^ PMID 25086476.
- ^ "Polycystic kidney disease". Mayo Clinic. Retrieved 2022-05-23.
- ISSN 0888-7543.
- ISSN 0888-7543.
- ISSN 1432-1203.
- PMID 10417277.
- PMID 9734362.
- PMID 12874107.
- PMID 14732716.
- S2CID 30757096.
- PMID 15569302.
- PMID 15780078.
- PMID 15677307.
- PMID 8559477.
- ^ PMID 25186187.
- PMID 21493772.
- PMID 17715262.
- PMID 12191984.
- PMID 11912216.
- PMID 19602418.
- PMID 18215695.
- PMID 21079243.
- PMID 15327388.
- ^ PMID 20219622.
- ^ PMID 25333066.
- PMID 22034641.
- PMID 20177400.
- S2CID 5777115.
- ^ "Autosomal Dominant Polycystic Kidney Disease". The Lecturio Medical Concept Library. Retrieved 3 July 2021.
- ^ PMID 35664270.
- PMID 34401721.
- ^ "Tolvaptan Cleared in US for ADPKD in Adults". 2018-04-26.
- PMID 10864573.
- PMID 24598801.
- ^ PMID 24904092.
- S2CID 22942772.
- ^ Brown T (2013). "Tolvaptan Not Recommended for ADPKD". Medscape.
- PMID 26764208.
- ^ PMID 31631001.
- ^ S2CID 204886698.
- PMID 32086281.
- S2CID 52033674.
- PMID 31631001.
- PMID 22207329.
- PMID 29118087.
- S2CID 219637514.
- S2CID 219637514.
- ^ PMID 31361604.
- PMID 27663039.
- S2CID 202733384.
- PMID 25534330.
- ^ PMID 16287460.
- ^ PMID 14511045.
- ^ PMID 8648810.
- ^ PMID 11206615.
- PMID 9162369.
- PMID 28159317.
- PMID 24436554.
- S2CID 199389236. Retrieved 5 January 2023.
- PMID 20219619.
- ^ PMID 17509331.
- ^ PMID 25210553.
- S2CID 25363382.
- PMID 18971106.
- PMID 19616833.
- ^ PMID 25852862.
- PMID 18300116.
- PMID 15201424.
- PMID 19825331.
- S2CID 27836789.
- PMID 23237695.
- ^ S2CID 22042874.
- )
External links
- "Polycystic Kidney Disease". National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). U.S. Department of Health and Human Services. Archived from the original on 2011-06-08.
- "Polycystic Kidney Disease". Genes and Disease [Internet]. Bethesda (MD): National Center for Biotechnology Information (US). 1998.