
Topoisomerase
| Identifiers | |||||||||
|---|---|---|---|---|---|---|---|---|---|
ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
| DNA Topoisomerase, ATP-dependent (type II) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
DNA topoisomerases (or topoisomerases) are enzymes that catalyze changes in the topological state of
Discovery
The first DNA topoisomerase was discovered in bacteria by James C. Wang in 1971 and was initially named ω (omega) protein;[3] it is now called Escherichia coli (E. coli) topoisomerase I (topo I) and is a representative of the type IA family of enzymes. Subsequently, a similar activity was found in eukaryotic cells (rat liver) by James Champoux and Renato Dulbecco;[4] the enzyme responsible, eukaryotic topo I, has a distinct mechanism and is representative of the type IB family. The first type II topoisomerase to be discovered was DNA gyrase from bacteria, by Martin Gellert and coworkers in 1976,[5] and also characterized by Nicholas Cozzarelli and co-workers.[6] DNA gyrase catalyzes the introduction of negative supercoils into DNA and is the only type II enzyme to do this, all the others catalyze DNA relaxation. Type II enzymes are mechanistically distinct from type I in being ATP-dependent and transiently cleaving both DNA strands rather than just one. Type II topoisomerases were subsequently identified from bacterial viruses and eukaryotes.[7][8][9] Topo EC-codes are as follows: ATP-independent (type I), EC 5.6.2.1; ATP-dependent (type II): EC 5.6.2.2. The exception among the type I topoisomerases, reverse gyrase, which contains a helicase domain (EC 3.6.4.12) and introduces positive supercoiling in an ATP-dependent manner. Therefore it is the sole type I topoisomerase classified as EC 5.6.2.2 (Table 1).
DNA topology
The double-helical structure of DNA involves the intertwining of the two polynucleotide strands around each other, which potentially gives rise to topological problems. DNA topology refers to the crossing of the two DNA strands that alters the twist of the double helix and gives rise to tertiary conformations of DNA, such as supercoils, knots and catenanes.[10] Potential topological issues associated with the double-helical structure of DNA were recognized soon after its structure was first elucidated in 1953 by James Watson, Francis Crick and Rosalind Franklin[11][12][13] and developed further by the work of Max Delbruck and John Cairns.[14][15] Closed-circular double-stranded DNA can be described by 3 parameters: Linking number (Lk), Twist (Tw) and Writhe (Wr) (Fig. 1). Where Lk refers to the number of times the two strands are linked, Tw refers to the number of helical turns in the DNA, measured relative to the helical axis, and Wr quantifies the coiling of the path of the DNA helix in space and is often equated with 'supercoiling'.
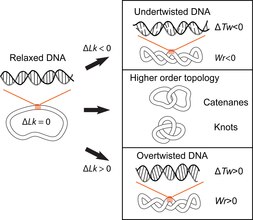
The 3 parameters are related as follows: Lk = Tw +Wr. This is a mathematical identity originally obtained by Călugăreanu in 1959[16] and is referred to as the Călugăreanu, or Călugăreanu–White–Fuller, theorem.[17][18] Lk cannot be altered without breaking one or both strands of the helix; Tw and Wr are interconvertible and depend upon the solution conditions. Supercoiling is a vernacular term for DNA with a non-zero linking difference, more correctly referred to as specific linking difference (σ = ΔLk/Lk0, where Lk0 is the mean linking number of the relaxed DNA circle). DNA is said to be positively supercoiled if Lk of it is higher than Lk0 for the relaxed state (Lk-Lko = ΔLk, ΔLk>0); that means that Tw and/or Wr are increased relative to the relaxed molecule. Conversely, DNA is negatively supercoiled if Lk of the molecule is lower than the Lk0 (ΔLk<0).
The consequences of topological perturbations in DNA are exemplified by DNA replication during which the strands of the duplex are separated; this separation leads to the formation of positive supercoils (DNA overwinding or overtwisting) ahead of the replication fork and intertwining of the daughter strands (precatenanes) behind[10][19] (Fig. 2). If the positive supercoils are not relaxed, progression of the replication fork is impeded, whereas failure to unlink the daughter strands prevents genome segregation, which is required for cell division.[20] Transcription by RNA polymerase also generates positive supercoiling ahead of, and negative supercoiling behind, the transcriptional complex (Fig. 2). This effect is known as the twin-supercoiled domain model, as described by Leroy Liu and James Wang in 1987.[21] These topological perturbations must be resolved for DNA metabolism to proceed, allowing the cell to efficiently replicate, transcribe and partition the genome to enable cellular division and vitality. Knots in DNA can be found in bacteriophages and as products of recombination reactions.[10] In general, knots in DNA are detrimental and need to be removed (by topoisomerases). DNA catenanes are formed upon the replication of circular molecules and need to be resolved by topoisomerases or recombinases to allow proper separation of daughter molecules during cell division. In addition to the detrimental aspects of DNA topology that require resolution, there are also beneficial aspects. For example, plasmid replication requires negative supercoiling of the origin, which facilitates local melting and exposes single-stranded DNA required to initiate replication. Similarly, initiation of replication from the main bacterial origin oriC also requires negative supercoiling.[22][23] Furthermore, compaction of the E. coli genome is achieved in part by negative supercoiling.
Types
DNA topoisomerases are enzymes that have evolved to resolve topological problems in DNA (Table 2).[10] They do this via transient breakage of one or both strands of DNA. This has led to the classification of topos into two types: type I, which catalyze reactions involving transient single-stranded breaks, and type II, which catalyze reactions involving transient double-stranded breaks (Fig. 3; Table 2). Sub-types exist within these classifications.

Type I
These enzymes catalyze changes in DNA topology via transient single-stranded breaks in DNA. Reactions can occur on both single- and double-stranded DNA substrates and can proceed via a 'swivel' or 'strand-passage' mechanism (Fig. 3). The range of reactions includes: DNA supercoil relaxation, unknotting of single-stranded circles, and decatenation, provided at least one partner has a single-stranded region. In the case of the archaeal enzyme, reverse gyrase, positive supercoiling of DNA is possible.[24]
Type IA
Type IA are monomeric and bind to single-stranded segments of DNA. They introduce a transient single-stranded break through the formation of a tyrosyl-phosphate bond between a tyrosine in the enzyme and a 5′-phosphate in the DNA. The segment of DNA within which the break occurs is called the 'gate' or G-segment, and its cleavage allows the passage of another segment of DNA, the 'transport' or T-segment, to be passed through in a 'strand-passage' process.

Type IB
Type IB topoisomerases catalyze reactions involving transient single-stranded breaks in DNA through the formation of a tyrosyl-phosphate bond between a tyrosine in the enzyme and a 3′-phosphate in the DNA. Rather than utilizing a strand-passage mechanism, these enzymes operate via a 'swivel' or 'controlled rotation' of the cleaved strand around the intact strand.
Type IC
Type IC topoisomerases share a similar mechanism to the type IB enzymes but are structurally distinct. The sole representative is topo V, found in the hyperthermophile Methanopyrus kandleri.[29]
Type II
Type II topoisomerases catalyze changes in DNA topology via transient double-stranded breaks in DNA. Reactions occur on double-stranded DNA substrates and proceed via a strand-passage mechanism (Fig. 5). The range of reactions include DNA relaxation, DNA supercoiling, unknotting, and decatenation. Whereas all type II topoisomerases can catalyze DNA relaxation, gyrase, an archetypal bacterial topoisomerase, can also introduce negative supercoils. In contrast to type I topoisomerases that are generally monomeric, type II topoisomerases are homodimers or heterotetramers. They are classified into two subtypes based on evolutionary, structural, and mechanistic considerations. The general strand-passage mechanism for the type II topos begins with the binding of one DNA duplex, termed the gate segment (G-segment), at the DNA gate. Another duplex, termed the transport segment (T-segment), is captured by an ATP-operated clamp and passed through a transient break in the G-segment, involving 5ʹ phosphotyrosine linkages in both strands, before it is released through the C-gate and the G-segment is re-ligated (Fig. 5). Enzyme turnover requires the binding and hydrolysis of ATP.

Type IIA
Type IIA topoisomerases catalyze transient double-stranded breaks in DNA through the formation of tyrosyl-phosphate bonds between tyrosines in the enzyme (one on each subunit) and 5′-phosphates staggered by 4 bases in opposite DNA strands. The strand-passage reaction can be intra- or intermolecular (Fig. 5), thus permitting changes in supercoiling and knotting, or unlinking, respectively. This process changes the linking number of the DNA by +/-2. Examples of type IIA topoisomerases include eukaryotic
Type IIB
Type IIB also catalyze transient double-stranded breaks through the formation of tyrosyl-phosphate bonds between tyrosines in the enzyme and 5′-phosphates in opposite strands of the DNA, but in the case of IIB enzymes the double-stranded breaks have a 2-base stagger. Type IIB enzymes show important structural differences, but are evolutionarily related to the type IIA enzymes. These differences include the lack of one of the protein 'gates' (the C gate) (Fig. 5). Originally found in archaea, they have also been found in eukaryotes, and, in particular, in plants; examples include topo VI and topo VIII. Topo VI is the best-studied enzyme of this sub-type and is thought to be a preferential decatenase.[31]
As drug targets
For the non-specialist perhaps the most important aspect of topoisomerases is their role as drug targets both for antibacterial and anti-cancer chemotherapy; several topoisomerase-targeted antibacterial and anti-cancer drugs are listed among the 2019 World Health Organization Model List of essential Medicines. The reason for this prominence is that their reactions proceed via transient breaks in DNA, which, if stabilized by drug binding, can lead to cell death due to the generation of toxic single- or double-stranded breaks in genomic DNA. The majority of topo-targeted drugs act in this way, i.e. they stabilize the enzyme-DNA covalent cleavage intermediate.[32][33][34]
Antibacterial compounds
Although type I topos, such as bacterial topo I, are viable antibiotic targets,[35] there are currently no compounds in clinical use that target these enzymes. However, the type II enzymes, DNA gyrase and DNA topoisomerase IV, have enjoyed enormous success as targets for the widely-used fluoroquinolone antibiotics, (Fig. 6).
Fluoroquinolones (FQs)
Quinolone antibacterial compounds were first developed in the 1960s and have been in clinical use since the 1980s.[36] FQ derivatives, such as ciprofloxacin, levofloxacin and moxifloxacin (Fig. 6) have been highly-successful. These compounds work by interacting with their target (gyrase or topo IV) and DNA at the cleavage site to stabilize the DNA-protein covalent cleavage intermediate. Specifically, they intercalate into the DNA and prevent the DNA religation step of the topoisomerase reaction (Fig. 5). This is a highly-effective mechanism of inhibition that is also used by several topoisomerase-targeted anti-cancer drugs. Despite their spectacular success, resistance to FQs is a serious problem.[36] A variety of other compounds, such as quinazolinediones and imidazolpyrazinones,[37] work in a similar manner and it is hoped that some of these will replace FQs in the future.
Aminocoumarins
Aminocoumarins (Fig. 6), such as novobiocin, clorobiocin and coumermycin A1, are natural products from Streptomyces that inhibit the ATPase reaction of gyrase and topo IV.[37] Although they can be very potent against their target, they suffer from permeability and toxicity issues, and thus have not enjoyed the level of clinical success of the FQs.
Proteinaceous inhibitors
There are a number of protein inhibitors of gyrase, including the bacterial toxins CcdB, MccB17, and ParE,[38][39][40] that stabilize the cleavage complex, in a similar manner to FQs. Although these proteins are not viable as antibacterials, their mode of action could inspire the development of novel antibacterial compounds. Other protein inhibitors of gyrase prevent DNA binding by the topoisomerase rather than stabilizing cleavage complexes. These include YacG[41] and pentapeptide repeat proteins, such as QnrB1 and MfpA;[42][43] these protein inhibitors also confer resistance to fluoroquinolones.

Anti-cancer compounds
Both human topo I and topo II (both α and β isoforms) can be targeted in anticancer chemotherapy (Fig. 7).[32][33][44][45][46][47] Most of these compounds act in a similar way to FQs, i.e. by stabilizing the DNA-protein covalent cleavage complex; for this they have become known as topoisomerase poisons, distinct from catalytic inhibitors.[33][34][48] Several human topoisomerase inhibitors are included on the World Health Organization's List of Essential Medicines.
Camptothecin (CPT)
Camptothecin (Fig. 7), originally derived from the tree
Etoposide (VP-16)
Etoposide (Fig. 7) and its close relative teniposide (VM-26) are epipodophyllotoxin derivatives obtained from the rhizome of wild mandrake that target topo II by stabilizing the covalent cleavage complex and preventing religation of the cleaved DNA.[46] These are typically used in conjunction with other chemotherapy drugs to treat cancers including testicular tumors, small-cell lung cancer, and leukemia. Etoposide treatment can result in secondary leukemias arising from specific genomic translocations, mainly involving topo IIβ.[46]
Doxorubicin

Doxorubicin (Fig. 7) and the related derivatives daunorubicin, epirubicin, and idarubicin are anthracyclines obtained from the bacterium Streptomyces[48] that target human topo II, stabilizing the cleavage complex in a similar manner to other topoisomerase poisons. Mitoxantrone is a synthetic anthracenedione that is chemically and functionally similar to anthracyclines.[47] The anthracyclines were the first topoisomerase inhibitors used to treat cancer and remain among the most widely employed and effective treatments for a broad range of cancers including breast cancer, lymphoma, leukemias, carcinomas, sarcomas, and other tumors.[47] These compounds are DNA intercalating agents and as such can impact a wide range of cellular DNA processes in addition to specifically poisoning topo II.[33] Additional cytotoxicity stems from redox reactions involving anthracyclines that generate reactive oxygen species. Generation of reactive oxygen, along with poisoning of topo IIβ, result in the dose-limiting cardiotoxicity of the anthracyclines.[33]
Merbarone
Merbarone is a thiobarbituric acid derivative, and dexrazoxane (ICRF-187), one of several related bisdioxopiperazine derivatives, (Fig. 7) are examples of catalytic inhibitors of topo II, i.e. they prevent completion of the catalytic cycle of topo II but do not stabilize the DNA cleavage complex. Whereas these catalytic inhibitors exhibit cytotoxicity and have been tested in clinical trials, they are not currently in clinical use for cancer therapy.[47] However, dexrazoxane, which blocks ATP hydrolysis by topo II, is used to prevent cardiotoxicity associated with the anthracyclines.[50][51]
| Topoisomerase | Subfamily type | Function | Multimericity | Metal dependence | ATP dependence | Single- or double-stranded cleavage? | Cleavage polarity | Change in link number (L) |
|---|---|---|---|---|---|---|---|---|
| Topoisomerase I
(E. coli) |
Type IA | Removes (-), but not (+) supercoils. Prevents excessive supercoiling of the genome, and supports transcription | Monomer | Yes (Mg2+) | No | SS | ||
| Topoisomerase III
(E. coli) |
Removes (-), but not (+) supercoils; overlapping function with topoisomerase IV | |||||||
| Topoisomerase IIIα
(H. sapiens) |
Removes (-), but not (+) supercoils; assists in the unlinking of precatenanes in cellular DNA replication; can catalyze the knotting, unknotting, and interlinking of single-stranded circles as well as the knotting, unknotting, catenation, and decatenation of gapped or nicked duplex DNA circles | |||||||
| Topoisomerase IIIβ
(H. sapiens) |
Has been shown to be a putative RNA topoisomerase. Involved in RNA processing | |||||||
| Reverse gyrase
(Archaea) |
Removes (-), but not (+) supercoils, introduces positive supercoils | Monomer and Heterodimer | Yes | |||||
| Topoisomerase I
(H. sapiens) |
Type IB | Removes (+) and (-) supercoils; supports fork movement during replication and transcription | Monomer | No | No | SS | 3' | ±n |
| Topoisomerase I
(Vaccinia virus) |
||||||||
| Topoisomerase V
(Archaea) |
Type IC | Relaxes (+) and (-) supercoils. Involved in DNA repair. | Monomer | No | No | SS | 3' | ±n |
| Topoisomerase II (DNA gyrase)
(E. coli) |
Type IIA | Generates (-) supercoils (the only topoisomerase known to do this) | Heterotetramer | Yes (Mg2+) | Yes | DS | 5' | ±2 |
| Topoisomerase IV
(E. coli) |
Decatenates replicated DNA; relaxes (+) supercoils faster than (-) | Heterotetramer | ||||||
| Topoisomerase IIα
(H. sapiens) |
Essential; Unlinks intertwined daughter duplexes in replication; contributes to DNA relaxation during transcription | Homodimer | ||||||
| Topoisomerase IIβ
(H. sapiens) |
Role in suppressing recombination or supporting transcription in neurons | Homodimer | ||||||
| Topoisomerase VI
(Archaea) |
Type IIB | Relaxes (+) and (-) supercoils; responsible for decatenating replication intermediates | Heterotetramer | Yes (Mg2+) | Yes | DS | 5' | ±2 |
Role of topoisomerase in transcriptional regulation
At least one topoisomerase, DNA topoisomerase II beta (topo IIβ), has a regulatory role in gene transcription. Topo IIβ–dependent double-strand DNA breaks and components of the DNA damage repair machinery are important for rapid expression of immediate early genes, as well as for signal-responsive gene regulation.[52][53][54][55] Topo IIβ, with other associated enzymes,[54] appears to be important for the release of paused RNA polymerase at highly transcribed or long genes.[56][57][58]
Topo IIβ in initiation of transcription
Stimulus-induced DNA double-strand breaks (DSBs) that are limited to a short-term (10 minutes to 2 hours) are induced by topo IIβ in the promoter regions of signal-regulated genes. These DSBs allow rapid up-regulation of expression of such signal responsive genes in a number of systems (see Table below). These signal-regulated genes include genes activated in response to stimulation with estrogen, serum, insulin, glucocorticoids (such as dexamethasone) and activation of neurons. When the induced DNA double-strand break has been repaired, then transcription of the signal-responsive gene returns to a low basal level.[52]

Topo IIβ and PARP-1 were found to be constitutively present at a moderate level near the transcription start site of a promoter of a signal-responsive gene. After the signal occurred, topo IIβ caused a double-strand break and PARP-1 was involved in replacing histone H1 by HMGB1/HMGA2, which can promote transcription.[55] Topo IIβ and PARP-1 increased at the site of the double-strand break and components of the non-homologous end joining DNA repair pathway, including DNA-PKcs, Ku70/Ku80 and DNA ligase IV assembled with topo IIβ and PARP-1. This assemblage was all present at the linker DNA adjacent to a single nucleosome in the promoter region of a gene (see Figure). The nucleosome was close to the transcription start site of the gene.[55] The components of the non-homologous end joining DNA repair pathway were essential to the closing of the DNA double-strand break.[52]
| Ligand or activating agent | Gene(s) evaluated for DSBs | DSB after signal-induced activation | DSB was required for transcription | DSB location | Proteins present at DSB | Duration of DSB after signal-induced activation | Refs |
|---|---|---|---|---|---|---|---|
| Estradiol | pS2 | yes | yes | promoter | 10 minutes | [59] | |
| Insulin | FASN | yes | yes | promoter | 3 hours | [60] | |
| Heat shock or Serum | yes | yes | promoter & POLII pausing site | DNA-PKcs, Ku70, γH2AX, TRIM28 | 30 seconds to 5 minutes | [61] | |
| Dexamethasone or Estradiol | PS2, MMTV, PLZF, HSD11B2 | yes | yes | promoter | , BRG1 | 15 minutes | [62] |
| KCl or NMDA activation of cultured primary cortical neurons | yes | yes | promoter | , CTCF | Up to 2 hours | [52] | |
| Fear conditioning (evaluated in mouse hippocampal and medial prefontal cortex neurons) | >200 genes with new DSBs and up-regulated expression | yes | DSBs were correlated with transcription | promoters | not tested | 10 minutes and second peak at 30 minutes | [63] |
Topo IIβ regulation of gene expression
RNA polymerase II frequently has a pausing site that is about 30–60 nucleotides downstream of the transcription start site of a gene.[64][65] The pausing of RNA polymerase II at these sites and the controlled release of the pausing is thought to have a regulatory role in gene transcription. As pointed out by Singh et al.,[58] "about 80% of highly expressed genes in HeLa cells are paused". Very short-term, but not immediately resealed, topo IIβ-induced DNA double-strand breaks occur at sites of RNA polymerase II pausing, and appear to be required for efficient release of the paused state and progression to gene transcription.[56][57][58] For the genes at which it occurs, the DNA double-stranded break induced by TOP2B is thought to be part of the process of regulation of gene expression.
See also
- DNA topology
- Supercoil
- Type I topoisomerase
- Type II topoisomerase
- Topoisomerase I
- Topoisomerase IIα
- Topoisomerase IIβ
- Topoisomerase IIIα
- Topoisomerase IIIβ
References
- ^ S2CID 231679533.
- PMID 33959387.
- PMID 4927945.
- PMID 4333036.
- PMID 186775.
- PMID 200930.
- S2CID 42645648.
- S2CID 4343962.
- PMID 226976.
- ^ OCLC 64239232.
- S2CID 4256010.
- S2CID 4253007.
- S2CID 4169572.
- PMID 14017761.
- PMID 16589559.
- ^ Călugăreanu G (1959). "L'intégrale de Gauss et l'analyse des nœuds tridimensionnels". Revue de Mathématiques Pure et Appliquées. 4: 5–20.
- PMID 5279522.
- JSTOR 2373348.
- PMID 11459956.
- S2CID 24408315.
- PMID 2823250.
- PMID 31266875.
- PMID 1488550.
- ^ S2CID 4242694.
- S2CID 8921868.
- PMID 17350929.
- ^ PMID 9488652.
- PMID 9136883.
- PMID 19106140.
- PMID 21525132.
- PMID 35076393.
- ^ PMID 23259582.
- ^ PMID 20534341.
- ^ PMID 22173432.
- PMID 25875873.
- ^ PMID 33271787.
- ^ OCLC 227210110.
- PMID 31181289.
- S2CID 40019620.
- PMID 16963775.
- PMID 24990966.
- PMID 33836580.
- PMID 33434265.
- PMID 6324188.
- PMID 8996602.
- ^ PMID 34008964.
- ^ ISBN 9780128125229.
- ^ PMID 9748560.
- PMID 19383846.
- PMID 3137469.
- PMID 15892593.
- ^ PMID 29254824.
- PMID 34610268.
- ^ S2CID 243846627.
- ^ S2CID 206508330.
- ^ S2CID 256819778.
- ^ PMID 31649282.
- ^ PMID 32029477.
- S2CID 206508330.
- PMID 19303849.
- PMID 26671524.
- PMID 26055322.
- PMID 34197463.
- PMID 33640324.
- PMID 29691322.
Further reading
- Wang JC (2009). Untangling the Double Helix: DNA entanglement and the action of the DNA topoisomerases. Cold Spring Harbor: Cold Spring Harbor Laboratory Press. p. 245. ISBN 978-0-87969-879-9.
External links
- DNA+Topoisomerases at the U.S. National Library of Medicine Medical Subject Headings (MeSH)