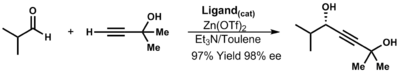Organozinc compounds were among the first organometallic compounds made. They are less reactive than many other analogous organometallic reagents, such as
protic solvents. For many purposes they are prepared in situ, not isolated, but many have been isolated as pure substances and thoroughly characterized.[6]
Organozincs can be categorized according to the number of carbon substituents that are bound to the metal.[2][3]
Diorganozinc (R2Zn): A class of organozinc compounds in which two alkyl ligands. These may be further divided into subclasses depending on the other
ligands
attached
Heteroleptic (RZnX): Compounds which an
electronegative or monoanionic ligand (X), such as a halide
, is attached to the zinc center with another alkyl or aryl substituent (R).
Ionic organozinc compounds: This class is divided into organozincates (RnZn−) and organozinc
cations
(RZnL+ n).
Bonding
In its
ligand field effects are nonexistent. Coordination geometry is thus determined largely by electrostatic and steric interactions.[2]
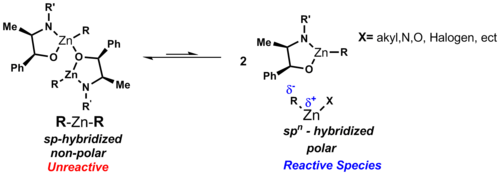
Organozinc compounds usually are two- or three-coordinate, reflecting the strongly donating property of the carbanionic ligands.
Typical diorganozinc complexes have the formula R2Zn. Dialkylzinc compounds are monomeric with a linear coordination at the zinc atom.
dipole moment of symmetric diorganozinc reagents can be seen as zero in these linear complexes, which explains their solubility in nonpolar solvents like cyclohexane. Unlike other binary metal alkyls, the diorganozinc species show a low affinity for complexation with ethereal solvent. Bonding in R2Zn is described as employing sp-hybridized orbitals on Zn.[2]
These structures cause zinc to have two bonding d-orbitals and three low-lying non-bonding d-orbitals (see
cluster chemistry). When a halogen ligand is added to the zinc atom both the acceptor and donor character of zinc is enhanced allowing for aggregation.[2]
Saturated diorganozinc reagents with bridging aryl groups
Synthesis
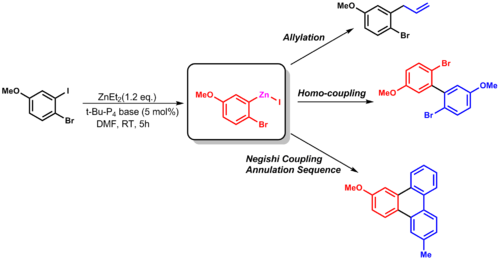
Several methods exist for the generation of organozinc compounds. Commercially available diorganozinc compounds are
Riecke zinc, produced by in situ reduction of ZnCl2 with potassium, is another activated form of zinc. This form has proven useful for reactions such as Negishi coupling and Fukuyama coupling. Formation of organozinc reagents is facilitated for alkyl or aryl halides bearing electron-withdrawing substituents, e.g., nitriles and esters.[10][11]
(2.3)
Using oxidative addition to get Negishi coupling precursors
(2.4)
Functional group exchange
The two most common zinc functional group interconversion reactions are with halides and boron, which is catalyzed by
diethyl zinc. This synthesis shows the utility of organozinc reagents by displaying high selectivity for the most reactive site in the molecule, as well as creating useful coupling partners.[12]
Organozinc function group exchange with metals or boron reagents
(2.5)
This group transfer reaction can be used in
allylation, or other coupling reactions (such as Negishi coupling).[13]
Hiroshi Naka and coworkers utilized this group transfer reaction to get to the key intermediate
(2.6)
β-Silyl diorganozinc compounds
One of the major drawbacks of diorganozinc alkylations is that only one of the two alkyl substituents is transferred. This problem can be solved by using Me3SiCH2- (TMSM), which is a non-transferable group.[14]
(2.7)
Transmetallation
Transmetallation is similar to the interconversions displayed above zinc can exchange with other metals such as mercury, lithium, copper, etc. One example of this reaction is the reaction of diphenylmercury with zinc metal to form diphenylzinc and metallic mercury
:
HgPh2 + Zn → ZnPh2 + Hg
(2.8)
The benefit of transmetalling to zinc it is often more tolerant of other functional groups in the molecule due to the low reactivity which increases selectivity.[15]
In the synthesis of Maoecrystal V, a
Esters are significantly stable against organozinc reagents.[16]
Zakarian's synthesis of Maoecrystal V utilized an early stage zinc transmetallation to tolerate functionality
(2.9)
Organozinc can be obtained directly from zinc metal:[17][18]
which quickly forms a soluble adduct with the organozinc compound thus removing it from the metal surface.
Reactions
In many of their reactions organozincs appear as intermediates.
In the Frankland–Duppa reaction (1863) an
alkyl halide R'X, zinc and hydrochloric acid to the α-hydroxycarboxylic esters RR'COHCOOR[19]
Reformatsky reaction
This organic reaction can be employed to convert α-haloester and ketone or aldehyde to a β-hydroxyester. Acid is needed to protonate the resulting alkoxide during work up. The initial step is an oxidative addition of zinc metal into the carbon-halogen bond, thus forming a carbon-zinc enolate. This C-Zn enolate can then rearrange to the Oxygen-Zinc enolate via coordination. Once this is formed the other carbonyl containing starting material will coordinate in the manner shown below and give the product after protonation.[20] The benefits of the Reformatsky reaction over the conventional aldol reaction protocols is the following:
Allows for exceedingly derivatized ketone substrates
The ester enolate intermediate can be formed in the presence of enolizable moieties
Below shows the six-membered transition state of the Zimmerman–Traxler model (Chelation Control, see Aldol reaction), in which R3 is smaller than R4.[21]
Basic mechanistic scheme of the Reformatsky reaction
(3.1)
The Reformatsky reaction has been employed in numerous total syntheses such as the synthesis of C(16),C(18)-bis-epi-cytochalasin D:[22]
E. Vedejs total synthesis of C(16),C(18)-bis-epi-cytochalasin D uses a late stage Reformatsky reaction to access the natural product
(3.2)
The Reformatsky reaction even allows for with zinc homo-enolates.[23] A modification of the Reformatsky reaction is the Blaise reaction.[21]
Scheme for the organozinc Blaise reaction, which utilizes an alpha-haloester and a functionalized cyano group
(3.3)
Simmons–Smith reaction
The
methylene iodide as the methylene source. The reaction is effected with zinc. The key zinc-intermediate formed is a carbenoid
(iodomethyl)zinc iodide which reacts with alkenes to afford the cyclopropanated product. The rate of forming the active zinc species is increased via ultrasonication since the initial reaction occurs at the surface of the metal.
(3.4)
The butterfly TS for the Barbier reaction
(3.5)
Although the mechanism has not been fully elaborated it is hypothesized that the organozinc intermediate is a metal-
Zinc-copper couple is commonly used to activate zinc.[21]
Directing groups aid in the selectivity of the Simmons–Smith reaction
propargyl. Alkylzinc coupling with alkyl halides such as bromides and chlorides have also been reported with active catalysts such as Pd-PEPPSI precatalysts, which strongly resist beta-hydride elimination (a common occurrence with alkyl substituents).[26] Either diorganic[check spelling] species or organozinc halides can be used as coupling partners during the transmetallation step in this reaction. Despite the low reactivity of organozinc reagents on organic electrophiles, these reagents are among the most powerful metal nucleophiles toward palladium.[27]
Alkylzinc species require the presence of at least a stoichiometric amount of halide ions in solution to form a "zincate" species of the form RZnX32−, before it can undergo transmetalation to the palladium centre.[28] This behavior contrasts greatly with the case of aryl zinc species. A key step in the catalytic cycle is a transmetalation in which a zinc halide exchanges its organic substituent for another halogen with the metal center.
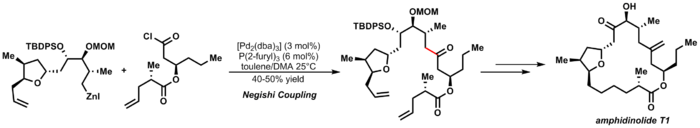
An elegant example of Negishi coupling is Furstner's synthesis of amphidinolide T1:[29]
Negishi Cross coupling reaction in the total synthesis of Amphidinolide T1
(3.7)
Fukuyama coupling
Fukuyama coupling is a palladium-catalyzed reaction involving the coupling of an aryl, alkyl, allyl, or α,β- unsaturated thioester compound. This thioester compound can be coupled to a wide range of organozinc reagents in order to reveal the corresponding ketone product. This protocol is useful due to its sensitivity to functional groups such as ketone, acetate, aromatic halides, and even aldehydes. The chemoselectivity observed indicates ketone formation is more facile than oxidative addition of palladium into these other moieties.[30]
Basic scheme for Fukuyama coupling of thioesters
(3.8)
A further example of this coupling method is the synthesis of (+)-biotin. In this case, the Fukuyama coupling takes place with the thiolactone:[31]
Total synthesis of (+)-biotin using Fukuyama coupling
(3.9)
Barbier reaction
The
1,2-addition. The Barbier reaction is advantageous because it is a one-pot process: the organozinc reagent is generated in the presence of the carbonyl substrate. Organozinc reagents are also less water sensitive, thus this reaction can be conducted in water. Similar to the Grignard reaction, a Schlenk equilibrium applies, in which the more reactive dialkylzinc can be formed.[21]
Basic scheme of the Barbier reaction
(3.10)
The mechanism resembles the
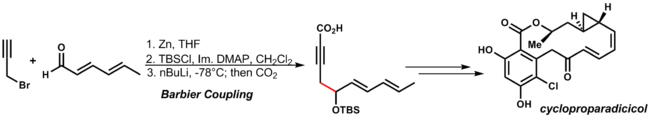
Danishefsky's synthesis of cycloproparadicicol. By using the organozinc addition reaction conditions the other functionality of the dienone and the alkyne are tolerated:[32]
Samuel Danashefskey's total synthesis of cycloproparadiciciol utilizes an early stage Barbier reaction to access the key intermediate.
alkali metals, such as sodium, and thus form these 'ate compounds'. Two types of organozincates are recognized: tetraorganozincates ([R4Zn]M2), which are dianionic, and triorganozincates ([R3Zn]M), which are monoanionic. Their structures, which are determined by the ligands, have been extensively characterized.[3]
Synthesis
Tetraorganozincates such as [Me4Zn]Li2 can be formed by mixing Me2Zn and MeLi in a 1:2 molar ratio of the rectants. Another example synthetic route to forming spriocyclic organozincates is shown below:[3]
Spirocyclic tetraorganozincate synthesis
(4.2)
Triorganozincates compounds are formed by treating a diorganozinc such as (Me3SiCH2)2Zn with an
alkali earth metal
(Ba, Sr, or Ca). One example is [(Me3SiCH2)3Zn]K.
Triethylzincate degrades to sodium hydridoethylzincate(II) as a result of
The product is an edge-shared bitetrahedral structure, with
hydride ligands
.
Reactions
Although less commonly studied, organozincates often have increased reactivity and selectivity compared to the neutral diorganozinc compounds. They have been useful in stereoselective alkylations of ketones and related carbonyls, ring opening reactions. Aryltrimethylzincates participate in vanadium mediated C-C forming reactions.[3]
One useful organozincate reaction
(4.4)
Organozinc(I) compounds
Low valent organozinc compounds having a Zn–Zn bond are also known. The first such compound, decamethyldizincocene, was reported in 2004.[40]





![{\displaystyle {\ce {{ZnCl2}+2K->[{\ce {THF}}][{\ce {-2KCl}}]}}\overbrace {\ce {Zn^{0}}} ^{\ce {Riecke\ zinc}}+{\ce {R-X->[{\ce {THF}}][20-60^{\circ }{\ce {C}}]R-Zn-I}}\qquad {\begin{cases}\mathbf {R} :&{\text{Allyl, Aryl, Alkyl, Benzyl}}\\\mathbf {X} :&{\text{Bromide, Iodide}}\end{cases}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/18c7cba23f482f24117a7664d614603a1271e9f2 "Reicke zinc allows for an activated zinc species for oxidative addition")





![{\displaystyle {\begin{array}{l}{}\\{\ce {{R2Zn}+(TMSM)2Zn}}\ {\overset {\ce {THF}}{\ce {<=>>}}}\ {\ce {2R(TMSM)Zn}}\\{}\\{\ce {{RZnI}+(TMSM)Li->[{\ce {THF}}][-80^{\circ }\!{\ce {C}}]{R(TMSM)Zn}+LiI}}\\{}\end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0fdd1941ffd29949716def994df7c380dd816d96 "Knochel and coworkers beta silyl group transfer addition")



















-Biotin_Fukuyama_Coupling.png)